
Строение молекулы HNO3 — Знаешь как
Атомы элементов второго периода не имеют свободных d-подуровней. Поэтому фтор проявляет только одну валентность, равную единице, кислород — двухвалентен, а азот — трехвалентен. Как же в таком случае объяснить строение молекул азотной кислоты HNO3? Приводимая обычно в учебниках структурная формула  согласно которой атом азота образует пять ковалентных связей, не совсем правильна. Строение молекулы HNO3 следует выражать формулой:
согласно которой атом азота образует пять ковалентных связей, не совсем правильна. Строение молекулы HNO3 следует выражать формулой:

Один электрон атома азота переходит в электронную оболочку атома кислорода. Возникший при этом ион N+

образует четыре ковалентные связи. Таким образом, в молекуле азотной кислоты азот действительно пятивалентен, однако одна его связь ионная, а другие четыре — ковалентные.
Структурные формулы совершенно равнозначны, но, поскольку из опыта известно, что два не соединенных с водородом атома кислорода в молекуле HNO3 неразличимы, каждая из этих формул в отдельности неверна. Лишь вместе они правильно отражают строение азотной кислоты. Чтобы показать это, пользуются знаком «резонанса»⇔,который обозначает,что истинное строение выражается некоторой промежуточной формулой:

Не нужно путать знак резонанса со знаком обратимости (⇄). Электроны не прыгают с одного атома кислорода на другой, чтобы «давать» то одну, то другую форму. Реальная молекула соответствует наложению крайних структур.
Поскольку электрон имеет корпускулярно-волновую природу, его можно считать как бы «размазанным» между, двумя атомами кислорода. Тогда вместо двух резонансных структур можно написать следующую формулу, отражающую строение молекулы азотной кислоты.

Статья на тему Строение молекулы HNO3
Cтроение молекулы оксида углерода с позиций методов ВС и МО. Задачи 955
Задача 955.
Описать свойства оксида углерода (II), указав: а) электронное строение молекулы с позиций методов ВС и МО; б) отношение к воде и к водным растворам кислот и щелочей; в) окислительно-восстановительные свойства.
Решение:
а) Электронное строение молекулы СО с позиций метода ВС. В атоме углерода имеется два неспаренных электрона на 2р-орбиталях, которые образуют связи по обычному ковалентному механизму с неспаренными р-электронами атома кислорода:
При этом образуется одна σ -связь и одна π -связь. У атома углерода одна р-орбиталь остаётся вакантной, и будет являться акцептором электронов при образовании связей по донорно-акцепторному механизму.
Электронное строение молекулы СО с позиций метода МО.
Схема заполнения молекулярных орбиталей молекулы СО:
Запись электронной конфигурации имеет вид:
1σn22σn24σn21πx21πу25σn2
Связывающий характер имеют три орбитали (4σn21πx21πу2 ) или как на схеме π2рх π2рz σ2рх), пеоэтому связь между атомами С и О является устойчивой и энергия её очень велика. Неподелённая электронная пара на 5σn
ω = (6 — 0)/2 = 3
Порядок связи 3 указывает на то, что связь в молекуле СО тройная, прочнее, чем обычная связь. Этим и объясняется значительное сходство СО с N2, — например, близость энергии диссоциации молекул (N2 — 945, СО – 1076 кДж/моль), межъядерных расстояний в молекулах (соответственно 0,110 и 0,113 нм), температур плавления (63 и 68 К) и кипения (77 и 82К). Отсутствие неспаренного электрона объясняет диамагнитные свойства молекулы СO. Диамагнитные свойства и термическую прочность СO с позиций метода ВС объяснить невозможно. Молекулу СО можно изобразить так:
б) Отношение СО к воде и водным растворам кислот и оснований.
СО плохо растворим в воде. СО – несолеобразующий оксид, не образует ни кислот, ни оснований. Хотя СО образуется при обезвоживании муравьиной кислоты и даёт при взаимодействии со щелочами её соли – фороиаты:
СО + NaOH HCOONa
его нельзя считать ангидридом этой кислоты, поскольку природа химической связи СО и НСООН различна. С кислотами СО не реагирует.
в) Окислительно-восстановительные свойства СО.
Оксид углерода (II) – сильный восстановитель. При высоких температурах он восстанавливает оксиды металлов, окисляясь до СО2. Например, в основе получения чугуна лежат реакции последовательного восстановления оксидов железа:
CO + 3Fe2O3 CO2 + 2Fe3O4;
CO + Fe
CO + FeO CO2 + Fe.
С некоторыми окислителями СО реагирует при небольшом нагревании:
В присутствии катализаторов СО окисляется до СО2:
2СО + О2 2СО2.
При взаимодействии СО с водородом, изменяя температуру, давление и катализатор, можно получить множество разнообразных продуктов: предельные углеводороды, спирты, альдегиды, кетоны, органические кислоты. Например:
СО + 2Н2 = СН3ОН.
Задача 956.
В каких случаях при горении угля образуется СО? Почему опасность появления угара при закрывании печи уменьшается по мере уменьшения накала углей? Для мотивировки ответа воспользоваться данными табличными данными относительно стандартных значений образования
Решение:
ΔG0298 (CO2) = 393,8 кДж/моль; ΔG0298 (CO) = 137,2 кДж/моль.
В промышленности СО получают при сжигании в определённых условиях. Реакция образования СО из угля описывается уравнением:
С + 1/2О2 = CO↑.
Стандартная энергия Гиббса этого процесса равна -137,2 кДж/моль. Реакция образования СО2 из угля описывается уравнением:
С + О2 = СО2↑.
Однако, стандартная
С + СО2 = 2СО↑.
Данная реакция эндотермична и при 298К изменение стандартной энергии Гиббса при её протекании положительна (+120, 4 кДж/моль). Однако в ходе превращения происходит двукратное увеличение числа молекул газа и энтропия системы увеличивается, так что энтропийное слагаемое энергии Гиббса будет иметь отрицательный знак. С увеличением температуры это слагаемое начинает преобладать (по абсолютной величине) над энтальпийным членом, в результате чего изменение
Таким образом, при высоких температурах уголь сгорает с образованием преимущественно СО (80%). Поэтому если процесс проводить при понижении температуры (или при уменьшении накала угля), а также при закрывании печи: в первом случае преобладает реакция:
С + О2 = CO2↑.
А во втором — протекает реакция:
2СО + О2 = 2CO2↑.
Естественно, при этих условиях уголь сгорает преимущественно до СО
Аммиак — Википедия
Аммиа́к (нитрид водорода, аммониак, гидрид азота) — бинарное неорганическое химическое cоединение азота и водорода с формулой Nh4{\displaystyle {\ce {Nh4}}}, при нормальных условиях — бесцветный газ с резким характерным запахом.
Плотность аммиака почти вдвое меньше, чем у воздуха, ПДКр.з. составляет 20 мг/м3 — IV класс опасности (малоопасные вещества) по ГОСТ 12.1.007-76. Растворимость Nh4{\displaystyle {\ce {Nh4}}} в воде чрезвычайно велика — около 1200 объёмов (при 0 °C) или 700 объёмов (при 20 °C) в объёме воды. В холодильной технике носит название R717, где R — Refrigerant (хладагент), 7 — тип хладагента (неорганическое соединение), 17 — молекулярная масса.
Аммиак относится к числу важнейших продуктов химической промышленности, ежегодное его мировое производство превышает 180 млн тонн.
Молекула аммиака имеет форму треугольной пирамиды с атомом азота в вершине. Три неспаренных p-электрона атома азота участвуют в образовании полярных ковалентных связей с 1s-электронами трёх атомов водорода (связи N−H{\displaystyle {\ce {N-H}}}), четвёртая пара внешних электронов является неподелённой, она может образовать ковалентную связь по донорно-акцепторному механизму с ионом водорода, образуя ион аммония Nh5+{\displaystyle {\ce {Nh5^+}}}. Несвязывающее двухэлектронное облако строго ориентировано в пространстве, поэтому молекула аммиака обладает высокой полярностью, что приводит к его хорошей растворимости в воде.
В жидком аммиаке молекулы связаны между собой водородными связями. Сравнение физических свойств жидкого аммиака с водой показывает, что аммиак имеет более низкие температуры кипения (tкип −33,35 °C) и плавления (tпл −77,70 °C), а также меньшие плотность, вязкость (в 7 раз меньше вязкости воды), проводимость (почти не проводит электрический ток) и диэлектрическую проницаемость. Это в некоторой степени объясняется тем, что прочность водородных связей в жидком аммиаке существенно ниже, чем у воды; а также тем, что в молекуле аммиака имеется лишь одна пара неподелённых электронов, в отличие от двух пар в молекуле воды, что не даёт возможность образовывать разветвлённую сеть водородных связей между несколькими молекулами. Аммиак легко переходит в бесцветную жидкость с плотностью 681,4 кг/м³, сильно преломляющую свет. Подобно воде, жидкий аммиак сильно ассоциирован, главным образом за счёт образования водородных связей. Жидкий аммиак — хороший растворитель для очень большого числа органических, а также для многих неорганических соединений. Твёрдый аммиак — кубические кристаллы.
- Благодаря наличию неподелённой электронной пары во многих реакциях аммиак выступает как основание Бренстеда или комплексообразователь. Так, он присоединяет протон, образуя ион аммония:
- Nh4+H+⟶Nh5+{\displaystyle {\ce {Nh4 + H^+ -> Nh5^+}}}.
- Водный раствор аммиака («нашатырный спирт») имеет слабощелочную реакцию из-за протекания процесса:
- Nh4+h3O⟶Nh5++OH−{\displaystyle {\ce {Nh4 + h3O -> Nh5^+ + OH^-}}}, Ko=1,8⋅10−5.
- Взаимодействуя с кислотами, даёт соответствующие соли аммония:
- Nh4+HNO3⟶Nh5NO3{\displaystyle {\ce {Nh4 + HNO3 -> Nh5NO3}}}.
- Аммиак также является очень слабой кислотой (в 10 000 000 000 раз более слабой, чем вода), способен образовывать с металлами соли — амиды, имиды и нитриды. Соединения, содержащие ионы Nh3−{\displaystyle {\ce {Nh3^-}}}, называются амидами, Nh3−{\displaystyle {\ce {NH^{2-}}}} — имидами, а N3−{\displaystyle {\ce {N^{3-}}}} — нитридами. Амиды щелочных металлов получают, действуя на них аммиаком:
- 2Nh4+2K⟶2KNh3+h3{\displaystyle {\ce {2Nh4 + 2K -> 2KNh3 + h3}}}.
Амиды, имиды и нитриды ряда металлов образуются в результате некоторых реакций в среде жидкого аммиака. Нитриды можно получить нагреванием металлов в атмосфере азота.
Амиды металлов являются аналогами гидроксидов. Эта аналогия усиливается тем, что ионы OH−{\displaystyle {\ce {OH^-}}} и Nh3−{\displaystyle {\ce {Nh3^-}}}, а также молекулы h3O{\displaystyle {\ce {h3O}}} и Nh4{\displaystyle {\ce {Nh4}}} изоэлектронны. Амиды являются более сильными основаниями, чем гидроксиды, а следовательно, подвергаются в водных растворах необратимому гидролизу:
- NaNh3+h3O⟶NaOH+Nh4{\displaystyle {\ce {NaNh3 + h3O -> NaOH + Nh4}}}.
и в спиртах:
- KNh3+C2H5OH⟶C2H5OK+Nh4{\displaystyle {\ce {KNh3 + C2H5OH -> C2H5OK + Nh4}}}.
Подобно водным растворам щелочей, аммиачные растворы амидов хорошо проводят электрический ток, что обусловлено диссоциацией:
- KNh3⇄K++Nh3−{\displaystyle {\ce {KNh3\rightleftarrows {K^{+}}+NH_{2}^{-}}}}.
Фенолфталеин в этих растворах окрашивается в малиновый цвет, при добавлении кислот происходит их нейтрализация. Растворимость амидов изменяется в такой же последовательности, что и растворимость гидроксидов: LiNh3{\displaystyle {\ce {LiNh3}}} — нерастворим, NaNh3{\displaystyle {\ce {NaNh3}}} — малорастворим, KNh3{\displaystyle {\ce {KNh3}}}, RbNh3{\displaystyle {\ce {RbNh3}}} и CsNh3{\displaystyle {\ce {CsNh3}}} — хорошо растворимы.
- При нагревании аммиак разлагается, проявляет восстановительные свойства. Так, он горит в атмосфере кислорода, образуя воду и азот. Окисление аммиака воздухом на платиновом катализаторе даёт оксиды азота, что используется в промышленности для получения азотной кислоты:
- 2Nh4→1200—1300 ∘CN2+3h3{\displaystyle {\ce {2Nh4->[1200{\text{—}}1300~^{\circ }{\text{C}}]{N2}+3h3}}} (реакция обратима),
- 4Nh4+3O2⟶2N2+6h3O{\displaystyle {\ce {4Nh4 + 3O2 -> 2N2 + 6h3O}}} (без катализатора, при повышенной температуре),
- 4Nh4+5O2⟶4NO+6h3O{\displaystyle {\ce {4Nh4 + 5O2 -> 4NO + 6h3O}}} (в присутствии катализатора, при повышенной температуре).
На восстановительной способности Nh4{\displaystyle {\ce {Nh4}}} основано применение нашатыря Nh5Cl{\displaystyle {\ce {Nh5Cl}}} для очистки поверхности металла от оксидов при их пайке:
- 3CuO+2Nh5Cl⟶3Cu+3h3O+2HCl+N2{\displaystyle {\ce {3CuO + 2Nh5Cl -> 3Cu + 3h3O + 2HCl + N_2}}}.
Окисляя аммиак гипохлоритом натрия в присутствии желатина, получают гидразин:
- 2Nh4+NaOCl⟶N2h5+NaCl+h3O{\displaystyle {\ce {2Nh4 + NaOCl -> N2h5 + NaCl + h3O}}}.
- Галогены (хлор, йод) образуют с аммиаком опасные взрывчатые вещества — галогениды азота (хлористый азот, иодистый азот).
- С галогеноалканами аммиак вступает в реакцию нуклеофильного присоединения, образуя замещённый ион аммония (способ получения аминов):
- Nh4+Ch4Cl⟶[Ch4Nh4]Cl{\displaystyle {\ce {Nh4 + Ch4Cl -> [Ch4Nh4]Cl}}} (гидрохлорид метиламмония).
- При 1000 °C аммиак реагирует с углём, образуя синильную кислоту HCN{\displaystyle {\ce {HCN}}} и частично разлагаясь на азот и водород. Также он может реагировать с метаном, образуя ту же самую синильную кислоту:
- 2Ch5+2Nh4+3O2⟶2HCN+6h3O{\displaystyle {\ce {2Ch5 + 2Nh4 + 3O2 -> 2HCN + 6h3O}}},
- Nh5OH⟶Nh4+h3O{\displaystyle {\ce {Nh5OH -> Nh4 + h3O}}}.
- C солями меди и с серебром образует комплексные соли-аммиакаты
- Cu(NO3)2+4Nh4⟶[Cu(Nh4)4](NO3)2{\displaystyle {\ce {Cu(NO3)2 + 4Nh4 -> [Cu(Nh4)4](NO3)2}}},
- Cu3(PO4)2+12Nh4⟶[Cu(Nh4)4]3(PO4)2{\displaystyle {\ce {Cu3(PO4)2 + 12Nh4 -> [Cu(Nh4)4]3(PO4)2}}},
- Cu(Ch4COO)2+4Nh4⟶[Cu(Nh4)4](Ch4COO)2{\displaystyle {\ce {Cu(Ch4COO)2 + 4Nh4 -> [Cu(Nh4)4](Ch4COO)2}}},
- AgNO3+2Nh4⟶[Ag(Nh4)2]NO3{\displaystyle {\ce {AgNO3 + 2Nh4 -> [Ag(Nh4)2]NO3}}}.
![{\displaystyle {\ce {AgNO3 + 2Nh4 -> [Ag(Nh4)2]NO3}}}](/800/600/https/upload.wikimedia.org/wikipedia/commons/thumb/1/1e/Ammoniak_Reaktor_BASF.jpg/220px-Ammoniak_Reaktor_BASF.jpg)
Аммиак был впервые выделен в чистом виде Дж. Пристли в 1774 году, который назвал его «щелочной воздух» (англ. alkaline air)[4]. Через одиннадцать лет, в 1785 году К. Бертолле установил точный химический состав аммиака[5]. С того времени в мире начались исследования по получению аммиака из азота и водорода. Аммиак был очень нужен для синтеза соединений азота, поскольку получение их из чилийской селитры ограничивалось постепенным истощением запасов последней. Проблема уменьшения запасов селитры обострилась к концу XIX века. Только в начале XX века удалось изобрести процесс синтеза аммиака, пригодный для промышленности. Это осуществил Ф. Габер, начавший трудиться над этой задачей в 1904 году и к 1909 году создавший небольшой контактный аппарат, в котором использовал повышенное давление (в соответствии с принципом Ле-Шателье) и катализатор из осмия. 2 июля 1909 года Габер устроил испытания аппарата в присутствии К. Боша и А. Митташа, оба — от Баденского анилинового и содового завода (BASF), и получил аммиак. К. Бош к 1911 году создал крупномасштабную версию аппарата для BASF, а затем был построен и 9 сентября 1913 года вступил в строй первый в мире завод по синтезу аммиака, который был расположен в Оппау (ныне район в черте города Людвигсхафен-на-Рейне) и принадлежал BASF. В 1918 году Ф. Габер стал лауреатом Нобелевской премии по химии «за синтез аммиака из составляющих его элементов». В России и СССР первая партия синтетического аммиака была получена в 1928 году на Чернореченском химическом комбинате[6].
Аммиак (в европейских языках его название звучит как «аммониак») своим названием обязан оазису Аммона в Северной Африке[источник не указан 169 дней], расположенному на перекрёстке караванных путей. В жарком климате мочевина (NH2)2CO, содержащаяся в продуктах жизнедеятельности животных, разлагается особенно быстро. Одним из продуктов разложения и является аммиак. По другим сведениям, аммиак получил своё название от древнеегипетского слова амониан[источник не указан 169 дней]. Так называли людей, поклоняющихся богу Амону. Они во время своих ритуальных обрядов нюхали минерал нашатырь (NH4Cl), который при нагревании испаряет аммиак[источник не указан 169 дней].
Жидкий аммиак, хотя и в незначительной степени, диссоциирует на ионы (автопротолиз), в чём проявляется его сходство с водой:
- 2Nh4⟶Nh5++Nh3−{\displaystyle {\ce {2Nh4 -> Nh5^+ + Nh3^-}}}.
Константа самоионизации жидкого аммиака при −50 °C составляет примерно 10−33 (моль/л)².
Жидкий аммиак, как и вода, является сильным ионизирующим растворителем, в котором растворяется ряд активных металлов: щелочные, щёлочноземельные, Mg{\displaystyle {\ce {Mg}}}, Al{\displaystyle {\ce {Al}}}, а также Eu{\displaystyle {\ce {Eu}}} и Yb{\displaystyle {\ce {Yb}}}. В отличие от воды с жидким аммиаком данные металлы не реагируют, а именно растворяются и могут быть выделены в исходном виде при испарении растворителя. Растворимость щелочных металлов в жидком Nh4{\displaystyle {\ce {Nh4}}} составляет несколько десятков процентов. В жидком аммиаке Nh4{\displaystyle {\ce {Nh4}}} также растворяются некоторые интерметаллиды, содержащие щелочные металлы, например, Na4Pb9{\displaystyle {\ce {Na4Pb9}}}.
Разбавленные растворы металлов в жидком аммиаке окрашены в синий цвет, концентрированные растворы имеют металлический блеск и похожи на бронзу. При испарении аммиака щелочные металлы выделяются в чистом виде, а щелочноземельные — в виде комплексов с аммиаком [Э(Nh4)6]{\displaystyle {\ce {[{\text{Э}}(Nh4)6]}}}, обладающих металлической проводимостью. При слабом нагревании эти комплексы разлагаются на металл и Nh4{\displaystyle {\ce {Nh4}}}.
Растворённый в Nh4{\displaystyle {\ce {Nh4}}} металл постепенно реагирует с образованием амида:
- 2Na+2Nh4⟶2NaNh3+h3{\displaystyle {\ce {2Na + 2Nh4 -> 2NaNh3 + h3}}}.
Получающиеся в результате реакции с аммиаком амиды металлов содержат отрицательный ион Nh3−{\displaystyle {\ce {Nh3^-}}}, который также образуется при самоионизации аммиака. Таким образом, амиды металлов являются аналогами гидроксидов. Скорость реакции возрастает при переходе от Li{\displaystyle {\ce {Li}}} к Cs{\displaystyle {\ce {Cs}}}. Реакция значительно ускоряется в присутствии даже небольших примесей h3O{\displaystyle {\ce {h3O}}}.
Металлоаммиачные растворы обладают металлической проводимостью, в них происходит распад атомов металла на положительные ионы и сольватированные электроны, окружённые молекулами Nh4{\displaystyle {\ce {Nh4}}}. Металлоаммиачные растворы, в которых содержатся свободные электроны, являются сильнейшими восстановителями.
Благодаря своим электронодонорным свойствам молекулы NH3 могут входить в качестве лиганда в комплексные соединения. Так, введение избытка аммиака в растворы солей d-металлов приводит к образованию их аминокомплексов:
- CuSO4+4Nh4⟶[Cu(Nh4)4]SO4{\displaystyle {\ce {CuSO4 + 4Nh4 -> [Cu(Nh4)4]SO4}}}.
- Ni(NO3)2+6Nh4⟶[Ni(Nh4)6](NO3)
Алкины — Википедия
Алки́ны (иначе ацетиленовые углеводороды) — углеводороды, содержащие тройную связь между атомами углерода, образующие гомологический ряд с общей формулой CnH2n-2. Атомы углерода при тройной связи находятся в состоянии sp-гибридизации.


Для алкинов характерны реакции присоединения. В отличие от алкенов, которым свойственны реакции электрофильного присоединения, алкины могут вступать также и в реакции нуклеофильного присоединения. Это обусловлено значительным s-характером связи и, как следствие, повышенной электроотрицательностью атома углерода. Кроме того, большая подвижность атома водорода при тройной связи обусловливает кислотные свойства алкинов в реакциях замещения.
Впервые ацетилен был получен в 1836 году Эдмундом Дэви, двоюродным братом знаменитого английского химика Гемфри Дэви, нагреванием уксуснокислого калия с древесным углем и последующей реакцией с водой образовавшегося карбида калия [1]. Дэви назвал свой газ «двууглеродистым водородом».
В 1862 году немецкий химик и врач Ф. Вёлер вновь открыл ацетилен, действуя водой на карбид кальция.
В 1863 году французский химик М. Бертло получил ацетилен, пропуская водород над раскалёнными электрической дугой графитовыми электродами[2]. Именно он дал газу имя ацетилен (от латинских слов acetum — уксус и греческого иле — дерево). Русское название «ацетилен» впервые было применено Д. И. Менделеевым[3].
Большую роль в изучении химии ацетилена и его производных в конце XIX века сыграл А. Е. Фаворский.
В 1895 году Ле Шателье обнаружил, что ацетилен, сгорая в кислороде, даёт очень горячее пламя, что впоследствии легло в основу ацетиленовой технологии сварки и резки тугоплавких металлов[4].
Простейшим алкином является этин (ацетилен C2H2). По номенклатуре IUPAC названия алкинов образуются от названий соответствующих алканов заменой суффикса «-ан» на «-ин»; положение тройной связи указывается арабскими цифрами.
Углеводородные радикалы, образованные от алкинов имеют суффикс «-инил», так CH≡C- называется «этинил».
Ниже представлены некоторые представители алкинов и их названия:

Различают внутреннюю тройную связь (пример: бут-2-ин) и концевую (пример: бут-1-ин).
Гомологический ряд алкинов:
- В противном случае, разница в положении тройной связи в двух разных молекулах алкинов (например, бутин-1 и пентин-2) будет сигнализировать о том, что эти вещества будут являтся структурными изомерами по положению связи.
У алкинов связь −С≡С− линейна (угол 180°) и находится в одной плоскости. Атомы углерода связаны одной σ- и двумя π-связями, максимальная электронная плотность которых расположена в двух взаимно перпендикулярных плоскостях[5]. Длина тройной связи примерно 0,121 нм, энергия связи 836 кДж/моль.

На представленной выше диаграмме приведены молекулярные орбитали этилена и ацетилена.
Алкины по своим физическим свойствам напоминают соответствующие алкены. Низшие (до С4) — газы без цвета и запаха, имеющие более высокие температуры кипения, чем аналоги в алкенах. Алкины плохо растворимы в воде, лучше — в органических растворителях.
| Физические свойства некоторых алкинов[6][7] | |||||
|---|---|---|---|---|---|
| № | Название | Формула | Т плавления,°С | Т кипения,°С | Плотность, d204 |
| 1 | Этин | С2H2 | −81,8 | −75 | 0,565* |
| 2 | Пропин | C3H4 | −101,5 | −23 | 0,670* |
| 3 | Бут-1-ин | HC≡C−CH2CH3 | −125,9 | 8,1 | 0,678* |
| 4 | Бут-2-ин | CH3−C≡C−CH3 | −32,3 | 27,0 | 0,694 |
| 5 | Пент-1-ин | HC≡C−C3H7 | −90,0 | 39,3 | 0,695 |
| 6 | Пент-2-ин | CH3−C≡C−C2H5 | −101,0 | 55,0 | 0,714 |
| 7 | 3-Метилбут-1-ин | HC≡C−CH(CH3)CH3 | н/д | 28,0 | 0,665 |
| 8 | Гекс-1-ин | HC≡C−C4H9 | −132,4 | 71,4 | 0,719 |
* Значения измерены при температуре кипения.
Нахождение в природе и физиологическая роль алкинов[править | править код]
В природе алкины практически не встречаются. В некоторых видах грибов Basidiomycetes были обнаружены в крайне малом количестве соединения, содержащие полиацетиленовые структуры[8].
Ацетилен обнаружен в атмосфере Урана[9], Юпитера[10] и Сатурна[11].
Алкины обладают слабым наркозным действием. Жидкие алкины вызывают судороги[12].
Основным промышленным способом получения ацетилена является электро- или термокрекинг метана, пиролиз природного газа и карбидный метод.
Карбидный метод (промышленный способ)[править | править код]
Прокаливанием в электрических печах смеси оксида кальция с коксом при 1800—2000°С получают карбид кальция:
CaO+3C→CaC2+CO{\displaystyle {\mbox{CaO}}+3{\mbox{C}}\rightarrow {\mbox{CaC}}_{2}+{\mbox{CO}}}
При действии на полученный карбид воды образуется гидроксид кальция и ацетилен:
CaC2+2h3O→C2h3+Ca(OH)2{\displaystyle {\mbox{CaC}}_{2}+2{\mbox{H}}_{2}{\mbox{O}}\rightarrow {\mbox{C}}_{2}{\mbox{H}}_{2}+{\mbox{Ca(OH)}}_{2}}
Пиролиз углеводородов (промышленный способ)[править | править код]
Суть способа заключается в пропускании над специальной огнеупорной насадкой смеси природного газа с воздухом, который, сгорая, поднимает температуру до 1500 °C. Затем на насадке происходит пиролиз метана[13]:
2Ch5→C2h3+3h3{\displaystyle 2{\mbox{CH}}_{4}\rightarrow {\mbox{C}}_{2}{\mbox{H}}_{2}+3{\mbox{H}}_{2}}
Крекинг природного газа (промышленный способ)[править | править код]
Электрокрекинг[править | править код]
Метод заключается в пропускании метана между двумя металлическими электродами с огромной скоростью. Температура 1500—1600°С. С химической точки зрения метод аналогичен методу пиролиза, отличаясь лишь технологическим и аппаратным исполнением[14].
Термоокислительный крекинг[править | править код]
В этом методе используется частичное окисление метана благодаря использованию теплоты, образующейся при его сгорании[14]:
6Ch5+4O2→C2h3+8h3+3CO+CO2+3h3O{\displaystyle 6{\mbox{CH}}_{4}+4{\mbox{O}}_{2}\rightarrow {\mbox{C}}_{2}{\mbox{H}}_{2}+8{\mbox{H}}_{2}+3{\mbox{CO}}+{\mbox{CO}}_{2}+3{\mbox{H}}_{2}{\mbox{O}}}
Метод прямого синтеза[править | править код]
Углерод напрямую взаимодействует с водородом при очень высоких температурах:
2C+h3→C2h3{\displaystyle 2{\mbox{C}}+{\mbox{H}}_{2}\rightarrow {\mbox{C}}_{2}{\mbox{H}}_{2}}
Этот метод имеет чисто историческое значение (получение ацетилена в 1863 году М. Бертло).
Электролиз солей непредельных карбоновых кислот[править | править код]
В 1864 году Кекуле получил ацетилен электролизом фумарата и малеата натрия[15]:
NaOOCCH=CHCOONa+2h3O→C2h3+2CO2+2NaOH+h3{\displaystyle {\mbox{NaOOCCH}}\!\!=\!\!{\mbox{CHCOONa}}+2{\mbox{H}}_{2}{\mbox{O}}\rightarrow {\mbox{C}}_{2}{\mbox{H}}_{2}+2{\mbox{CO}}_{2}+2{\mbox{NaOH}}+{\mbox{H}}_{2}}
Аналогично получается ацетилен и из акрилата натрия.
Этот метод носит чисто историческое значение.
Дегидрогалогенирование галогеналканов и галогеналкенов (лабораторный способ)[править | править код]
Реакция дегидрогалогенирования проводят действием сильного основания на дигалогеналканы:

В качестве дегидрогалогенирующего агента удобно использовать амид натрия в жидком аммиаке[16]:

Алкилирование алкинов (лабораторный способ)[править | править код]
Алкилирование алкинов с концевой тройной связью производится по следующей схеме:

Подробнее смотри подраздел: Реакции нуклеофильного замещения алкинидов.
Прочие лабораторные способы получения алкинов[править | править код]
На первой стадии идёт образование дибромалкена:

На второй стадии происходит обмен брома на литий и альфа-элиминирование с последующим превращением винилидена в алкин в результате перегруппировки Фрича-Буттенбергера-Вихеля:
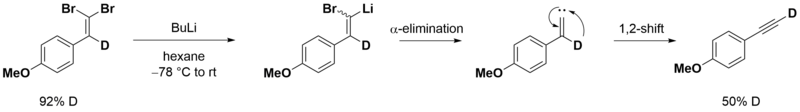
- Разложение дигидразонов[18]:

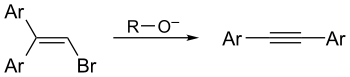
- Перегруппировка Фрича-Буттенберга-Вихелля — превращение 1,1-диарил-2-дигалогенэтиленов в производные ацетилена под действием сильных оснований[19]:
Кислотные свойства алкинов и реакции нуклеофильного замещения[править | править код]
Образование алкинидов[править | править код]
Алкины с концевой тройной связью являются С-H кислотами (сильнее чем аммиак и алкены, но слабее, чем спирты) которые с очень сильными основаниями могут образовывать соли — алкиниды[6]:
HC≡CH+2NaNh3→NaC≡CNa+2Nh4{\displaystyle {\mbox{HC}}\!\!\equiv \!\!{\mbox{CH}}+2{\mbox{NaNH}}_{2}\rightarrow {\mbox{NaC}}\!\!\equiv \!\!{\mbox{CNa}}+2{\mbox{NH}}_{3}} (ацетиленид динатрия)
HC≡CH+2K→KC≡CK+h3{\displaystyle {\mbox{HC}}\!\!\equiv \!\!{\mbox{CH}}+2{\mbox{K}}\rightarrow {\mbox{KC}}\!\!\equiv \!\!{\mbox{CK}}+{\mbox{H}}_{2}} (ацетиленид дикалия)
Ch4−C≡CH+C2H5MgBr→Ch4−C≡CMgBr+C2H6{\displaystyle {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{CH}}+{\mbox{C}}_{2}{\mbox{H}}_{5}{\mbox{MgBr}}\rightarrow {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{CMgBr}}+{\mbox{C}}_{2}{\mbox{H}}_{6}} (пропинилмагнийбромид)
Реакция алкинов с аммиакатами серебра или одновалентной меди является качественной на наличие концевой тройной связи[6]:
Ch4−C≡CH+Ag(Nh4)2OH→Ch4−C≡C−Ag↓+ 2Nh4+h3O{\displaystyle {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{CH}}+{\mbox{Ag(NH}}_{3})_{2}{\mbox{OH}}\rightarrow {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{C}}\!\!-\!\!{\mbox{Ag}}\!\downarrow +\ 2{\mbox{NH}}_{3}+{\mbox{H}}_{2}{\mbox{O}}}
Ch4−C≡CH+Cu(Nh4)2OH→Ch4−C≡C−Cu↓+ 2Nh4+h3O{\displaystyle {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{CH}}+{\mbox{Cu(NH}}_{3})_{2}{\mbox{OH}}\rightarrow {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{C}}\!\!-\!\!{\mbox{Cu}}\!\downarrow +\ 2{\mbox{NH}}_{3}+{\mbox{H}}_{2}{\mbox{O}}}
HC≡CH+2Cu(Nh4)2OH→Cu−C≡C−Cu↓+ 4Nh4+2h3O{\displaystyle {\mbox{HC}}\!\!\equiv \!\!{\mbox{CH}}+2{\mbox{Cu(NH}}_{3})_{2}{\mbox{OH}}\rightarrow {\mbox{Cu}}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{C}}\!\!-\!\!{\mbox{Cu}}\!\downarrow +\ 4{\mbox{NH}}_{3}+2{\mbox{H}}_{2}{\mbox{O}}}
Пропинид серебра представляет собой осадок белого цвета, пропинид меди — осадок жёлтого цвета, наконец, диацетиленид меди — осадок красного цвета.
Алкинид серебра легко растворяется при добавлении цианида натрия с выделением соответствующего алкина[8]:
Ch4−C≡C−Ag+2NaCN+h3O→Ch4−C≡CH↑+Na[Ag(CN)2]+NaOH{\displaystyle {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{C}}\!\!-\!\!{\mbox{Ag}}+2{\mbox{NaCN}}+{\mbox{H}}_{2}{\mbox{O}}\rightarrow {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{CH}}\!\uparrow +{\mbox{Na}}[{\mbox{Ag(CN)}}_{2}]+{\mbox{NaOH}}}
См. также статью: Ацетилениды.
Реакции нуклеофильного замещения алкинидов[править | править код]
Алкиниды являются сильными нуклеофилами и легко вступают в реакции нуклеофильного замещения:
NaC≡CNa+2h3O→HC≡CH+2NaOH{\displaystyle {\mbox{NaC}}\!\!\equiv \!\!{\mbox{CNa}}+2{\mbox{H}}_{2}{\mbox{O}}\rightarrow {\mbox{HC}}\!\!\equiv \!\!{\mbox{CH}}+2{\mbox{NaOH}}}
Это, в частности, широко используется для синтеза гомологов ацетилена:
HC≡CK+C4H9Br→HC≡C−C4H9+KBr{\displaystyle {\mbox{HC}}\!\!\equiv \!\!{\mbox{CK}}+{\mbox{C}}_{4}{\mbox{H}}_{9}{\mbox{Br}}\rightarrow {\mbox{HC}}\!\!\equiv \!\!{\mbox{C}}\!\!-\!\!{\mbox{C}}_{4}{\mbox{H}}_{9}+{\mbox{KBr}}}
Получение алкингалогенидов[править | править код]
Действием галогена на монозамещенные ацетилены в щелочной среде можно получить галогеналкины[14]:
Ch4−C≡CH+Br2+NaOH→Ch4−C≡C−Br+NaBr+h3O{\displaystyle {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{CH}}+{\mbox{Br}}_{2}+{\mbox{NaOH}}\rightarrow {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{C}}\!\!-\!\!{\mbox{Br}}+{\mbox{NaBr}}+{\mbox{H}}_{2}{\mbox{O}}}
Реакция нуклеофильного замещения алкинидов[править | править код]
Ch4−C≡C−MgBr+Br−Ch3−CH=Ch3→Ch4−C≡C−Ch3−CH=Ch3+MgBr2{\displaystyle {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{C}}\!\!-\!\!{\mbox{MgBr}}+{\mbox{Br}}\!\!-\!\!{\mbox{CH}}_{2}\!\!-\!\!{\mbox{CH}}\!\!=\!\!{\mbox{CH}}_{2}\rightarrow {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{C}}\!\!-\!\!{\mbox{CH}}_{2}\!\!-\!\!{\mbox{CH}}\!\!=\!\!{\mbox{CH}}_{2}+{\mbox{MgBr}}_{2}}
В препаративном синтезе часто используют комплекс ацетиленида лития с этилендиамином как удобный источник ацетиленид-аниона[8].
Следует отметить, что в случае реакции с вторичными или третичными галогеналканами реакция во многом идёт по альтернативному пути (элиминирование):
Ch4−C≡C−MgBr+Ch4−CHBr−Ch4→Ch4−C≡CH+Ch4−CH=Ch3{\displaystyle {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{C}}\!\!-\!\!{\mbox{MgBr}}+{\mbox{CH}}_{3}\!\!-\!\!{\mbox{CHBr}}\!\!-\!\!{\mbox{CH}}_{3}\rightarrow {\mbox{CH}}_{3}\!\!-\!\!{\mbox{C}}\!\!\equiv \!\!{\mbox{CH}}+{\mbox{CH}}_{3}\!\!-\!\!{\mbox{CH}}\!\!=\!\!{\mbox{CH}}_{2}}
Прочие реакции[править | править код]
Хлорированием ацетилена хлоридом меди (II) в водных растворах CuCl можно получить дихлорацетилен[20]:
HC≡CH+4CuCl2→ClC≡CCl+4CuCl+2HCl{\displaystyle {\mbox{HC}}\!\!\equiv \!\!{\mbox{CH}}+4{\mbox{CuCl}}_{2}\rightarrow {\mbox{ClC}}\!\!\equiv \!\!{\mbox{CCl}}+4{\mbox{CuCl}}+2{\mbox{HCl}}}
Ацетиленовая конденсация[править | править код]
Ацетиленовая конденсация или иначе реакция Ходкевича-Кадио, заключается во взаимодействии ацетиленовых углеводородов с бром- или йодалкинами с образованием диацетиленов[21]:

Аналогично протекает и реакция Куртца (катализатор — ацетиленид меди):
Ch3=CH−Ch3Cl+RC≡CH+NaOH→Ch3=CH−Ch3−C≡CR+NaCl+h3