
ФИЗИКО-ХИМИЯ ПОВЕРХНОСТНЫХ ЯВЛЕНИЙ — TDMUV
ФИЗИКО-ХИМИЯ ПОВЕРХНОСТНЫХ ЯВЛЕНИЙ
Причины возникновения поверхностных явлений
Поверхностные явления – это процессы, возникающие на любой границе раздела двух или нескольких фаз и приводящие к изменению свойств веществ при переходе от протяженного тела к межфазным поверхностным слоям.
Эти явления
обусловлены тем , что
контактирующие фазы различаются по структуре и, соответственно, по
физико-химическим свойствам. Следовательно, силовое поле, действующее на структурные
элементы вещества (молекулы, ионы, атомы, атомные группировки, ассоциаты),
которые находятся в поверхностных слоях и контактируют с другими фазами, будет
отличаться от силового поля, которое действует на структурные элементы вещества
в объеме тела. И чем больше различия в свойствах фаз, тем сильнее будут
выражены различия в свойствах межфазной поверхности и объема каждой из фаз.
Наиболее наглядно
представить поверхностные явления можно на примере границы раздела жидкость/газ.
В объеме жидкости на каждую молекулу действуют одинаковые по величине силы
взаимного притяжения со стороны соседних молекул, т. е. силовое поле
симметрично. Поэтому средние во времени равнодействующие этих сил равны нулю. Молекулы,
расположенные в поверхностном слое жидкости, испытывают притяжение со стороны
находящихся под ними «объемных» молекул, но это притяжение компенсируется лишь
частично их притяжением к молекулам газовой среды из-за низкой плотности
воздуха и гидрофобности его молекул. Поэтому для поверхностных молекул жидкости
равнодействующая молекулярных сил не равна нулю и направлена внутрь жидкости.
Иными словами, молекулы в поверхностном слое находятся в двумерном
энергетически нескомпенсированном состоянии в отличие от трехмерного (компенсированного)
состояния молекул в объеме. Таким образом, молекулы поверхностного слоя обладают
избыточной потенциальной энергией (отметим, что энергия частиц поверхностного
слоя в некоторых случаях может быть и меньше средней энергии частиц в объеме).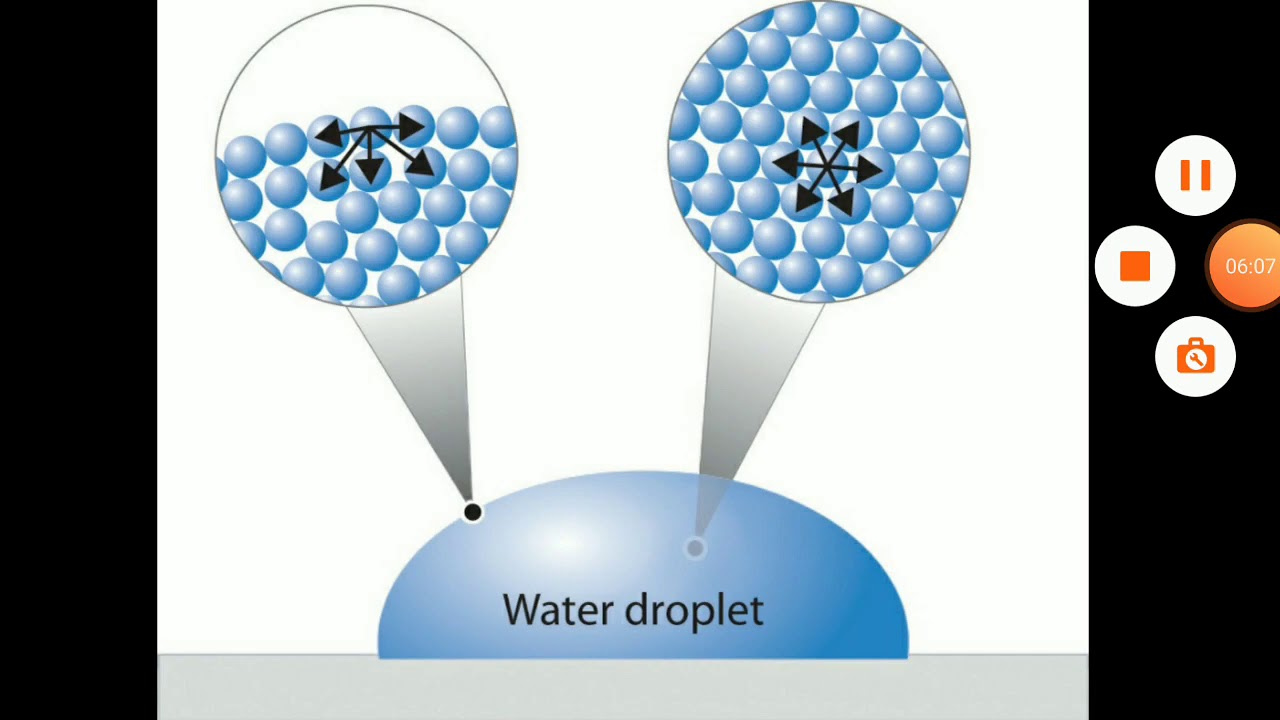
Многие процессы – испарение, сублимация и конденсация, адсорбция, диффузия, гетерогенный катализ, химические реакции в гетерогенных системах – протекают на границе раздела фаз. В этих процессах вещество либо переходит через поверхностный слой, либо поглощается им, либо вытесняется из него в объем. С поверхностными свойствами веществ связаны также процессы смачивания, трения, смазочного действия, адгезии.
Вы уже отлично знаете, что наличие высокоразвитой поверхности раздела фаз – это принципиальная особенность высокодисперсных систем. Влияние поверхностей раздела фаз и связанных с ними поверхностных явлений на свойства дисперсных систем обусловлено избыточной поверхностной энергией
Молекулы
конденсированных фаз, находящиеся в поверхности раздела, обладают избыточной
энергией по сравнению с молекулами в объеме из-за нескомпенсированности их
межмолекулярных взаимодействий. (Проиллюстрируйте эту фразу схематически!) Нескомпенсированность
межмолекулярных взаимодействий, в свою очередь, обусловлено различием состава и
строения контактирующих фаз. Это порождает возникновение на поверхности раздела
поверхностных сил и избытка энергии – поверхностной энергии.
(Проиллюстрируйте эту фразу схематически!) Нескомпенсированность
межмолекулярных взаимодействий, в свою очередь, обусловлено различием состава и
строения контактирующих фаз. Это порождает возникновение на поверхности раздела
поверхностных сил и избытка энергии – поверхностной энергии.
Наиболее просто связь между поверхностными свойствами и термодинамическими характеристиками контактирующих фаз проявляется для однокомпонентных двухфазных систем (например, жидкость на границе с ее насыщенным паром).
На поверхности
раздела фаз молекулы испытывают сильное притяжение со стороны жидкой фазы и
почти никакого притяжения со стороны паровой фазы. Равнодействующая сил
направлена в сторону жидкости и стремится втянуть молекулы внутрь жидкой фазы.
Эта равнодействующая сила называется внутренним молекулярным давлением. Его причиной
является межмолекулярное взаимодействие, и чем оно больше, тем больше
внутреннее давление. Так, для воды оно равно 15∙10 Вы видите, таким образом, что
для обеих жидкостей внутреннее давление довольно велико. Именно поэтому
жидкости малосжимаемы.
Вы видите, таким образом, что
для обеих жидкостей внутреннее давление довольно велико. Именно поэтому
жидкости малосжимаемы.
Поверхностное натяжение
Две вышеупомянутые фазы (жидкость и пар) могут существовать в равновесии только при наличии устойчивой границы раздела между ними, не проявляющей тенденции к самопроизвольному увеличению, т. е. термодинамически устойчивой (при данных температуре и объеме системы). С макроскопической точки зрения это означает, что с поверхностью связана некоторая энергия, так что общая энергия системы не является суммой энергии двух объемных фаз, а включает еще и избыточную энергию, пропорциональную площади поверхности раздела фаз:
.
Входящая в это
уравнение величина σ – это удельная (приходящаяся на единицу площади) поверхностная
энергия. Наличие на поверхности раздела фаз избытка энергии означает, что для
образования новой поверхности нужно совершить работу, поэтому величина σ
одновременно представляет собой работу обратимого изотермического образования
единицы поверхности; эту величину также называют поверхностным натяжением.
Поверхностное натяжение можно трактовать и как силу, действующую тангенциально к поверхности раздела фаз и препятствующую ее увеличению.
Отметим, что понятие «поверхностное натяжение» было введено голландскими физиками значительно ранее, чем появилось понятие «энергия» и означало силу, стягивающую гипотетическую пленку (аналогичную эластичной пленке) на поверхности жидкости и противодействующую ее растяжению.
В поверхностном слое действуют только межмолекулярные силы, и поэтому проводимую аналогию поверхностного слоя с эластичной пленкой нельзя признать полной, так как при растяжении такой пленки сила возрастает пропорционально деформации (по закону Гука), в то время как для однофазной границы жидкость-газ независимо от величины площади пленки σ=const.
Известно, что при
повышении температуры поверхностное натяжение чистых жидкостей уменьшается
приблизительно по прямолинейному закону, т. е. и только вблизи
критической температуры эта зависимость отклоняется от прямолинейной. Отметим,
что для индивидуальных веществ, например, для воды без примесей, когда
межмолекулярное взаимодействие и формирование поверхностного слоя
осуществляется одними и теми же молекулами, значения поверхностного натяжения в
энергетическом и силовом выражении (их обозначают единым символом σ)
совпадают.
Отметим,
что для индивидуальных веществ, например, для воды без примесей, когда
межмолекулярное взаимодействие и формирование поверхностного слоя
осуществляется одними и теми же молекулами, значения поверхностного натяжения в
энергетическом и силовом выражении (их обозначают единым символом σ)
совпадают.
Для растворов, в которых межмолекулярное взаимодействие их компонентов (растворенных веществ и растворителя) различно, значения поверхностного натяжения в виде энергии и силы не совпадают.
, ,
для индивидуальных веществ , а для растворов .
Для твердых
веществ применение понятий «поверхностное натяжение» и «удельная поверхностная
энергия» следует рассматривать с учетом того, что в этом случае работа,
затрачиваемая на образование новой поверхности, будет неодинакова в различных
местах. Неоднородность поверхности и ограниченная подвижность молекул этих тел
будут сказываться на поверхностном натяжении и следует его рассматривать или в
определенной точке поверхности или как усредненное значение.
Описание поверхностных явлений
В гетерогенных системах различают два типа межмолекулярного взаимодействия: внутрифазное и межфазное. Притяжение атомов и молекул внутри отдельной фазы называют
К межфазным взаимодействиям относятся понятия адгезии, смачивания и растекания. Адгезия (от лат. adhaesio – прилипание) – взаимодействие между приведенными в контакт поверхностями конденсированных фаз разной природы. Адгезия обеспечивается физическими или химическими силами взаимодействия. Смачивание и растекание – это адгезионные взаимодействия между жидким и твердым телом.
Явления адгезии и
смачивания широко распространены в природе и технологических процессах:
склеивание различных материалов, нанесение лаков, красок, металлических
покрытий, печать, крашение тканей, изготовление различных материалов на основе
связующих веществ (стеклопластики, резина, бетон и т.
Чтобы разорвать столбик жидкости с образованием двух новых поверхностей раздела , необходимо затратить работу, которая равна работе когезии, но противоположна по знаку:
Wразр = –dG
Изменение энергии Гиббса в этом процессе:
dG = dGкон – dGнач = s12 + s12 – 0 = 2s12
–Wразр = Wк
Wк = 2s12
Работа, затрачиваемая на разрыв поверхностного слоя, будет равна работе адгезии с обратным знаком:
–Wразр = dG,
где dG – изменение энергии Гиббса в
процессе разрушения адгезионного слоя.
dGнач = s13
dGкон = s32 + s12 – s13
dG = dGкон – dGнач = s32 + s12 – s13
–Wразр = Wа = dG
Wа = s32 + s12 – s13 (уравнение Дюпре)
Рассмотрите на качественном уровне возможные варианты поведения капли жидкости на поверхности твердого тела.
Капля на поверхности твердого тела представляет собой трехфазную систему: фаза Ж – жидкость капли, фаза Г – воздух, фаза Т – твердое тело
Условие
растекания капли: sТГ > sТЖ.
Следует также принять во внимание, что в этом процессе увеличивается энергия на границе жидкость–газ и поэтому можно ожидать остановку процесса растекания при достижении равенства sТГ – sТЖ и sЖГ·cosθ, или неограниченное растекание, если sТГ – sТЖ > sЖГ·cosθ.
Если sТГ < sТЖ может произойти стягивание капли. В первом случае происходит смачивание, а во втором – несмачивание твердого тела жидкостью.
Уравнение, выражающее взаимосвязь работы когезии, поверхностного натяжения и краевого угла смачивания — это уравнение называется уравнением Юнга-Дюпре:
.
Таким образом, смачивание, характеризующееся косинусом краевого угла θ, определяется отношением работы адгезии к работе когезии для смачивающей жидкости.
Рассмотренные
выше закономерности смачивания выполняются не на всех поверхностях, а только на
идеально гладких и однородных. Реально же твердые тела всегда имеют
неоднородности, от которых зависит краевой угол смачивания, и которые
затрудняют определение краевых углов.
Реально же твердые тела всегда имеют
неоднородности, от которых зависит краевой угол смачивания, и которые
затрудняют определение краевых углов.
Предположим, что
мы опустили капилляр в некую жидкость. Как Вы уже знаете из материала
предыдущей лекции, в зависимости от природы жидкости и материала капилляра
возможны два варианта: жидкость смачивает капилляр, и жидкость не смачивает
капилляр. В первом случае мы будем наблюдать самопроизвольное поднятие уровня
жидкости по капилляру на некоторую величину. Это обусловлено тем, что
поверхностно натяжение на границе ТГ больше, чем геометрическая сумма сил,
обусловленная действием поверхностного натяжения на границах ТЖ и ЖГ: . В результате система, проявляя тенденцию к снижению общей
ее энергии, стремится к уменьшению sТГ путем покрытия как можно большей
площади твердого тела жидкостью (при этом площадь границы ТГ уменьшается и
соответственно уменьшается энергия Гиббса, равная sS). Иными словами, сила взаимодействия жидкости с материалом
капилляра преобладает над суммарной силой взаимодействия жидкости с газом и
газа с материалом капилляра.
Иными словами, сила взаимодействия жидкости с материалом
капилляра преобладает над суммарной силой взаимодействия жидкости с газом и
газа с материалом капилляра.
В противоположном случае, когда имеет место обратное неравенство , уровень жидкости в капилляре становится ниже уровня жидкости в сосуде.
В результате описанных процессов мы будем наблюдать либо вогнутый, либо выпуклый (не плоский!) мениск жидкости в капилляре, т. е. форма межфазной поверхности на границе ЖГ будет искривлена (в данном случае она будет сферической).
Искривление межфазной поверхности наблюдается практически для любых систем на микроуровне. Идеально плоская поверхность наблюдается в редких случаях и является скорее исключением. Однако сильно выраженное искривление характерно именно для высокодисперсных систем, т. е. для мелких частиц – частиц дисперсной фазы золей, суспензий, эмульсий, нитей, капелек жидкости в газе и т. п.
Представим себе
какую-нибудь систему, в которой имеется искривленная поверхность. Для
наглядности возьмем мыльный пузырь, выдуваемый через трубочку. Если, выдув
пузырь, оставить трубочку открытой, пузырь тут же сдуется. Это свидетельствует
о том, что давление с внешней стороны пузыря превышает давление внутри пузыря.
Иными словами, в состоянии равновесия давления в фазах, разделенных
искривленной поверхностью, различаются. Эта разность давлений называют капиллярным
давлением.
Для
наглядности возьмем мыльный пузырь, выдуваемый через трубочку. Если, выдув
пузырь, оставить трубочку открытой, пузырь тут же сдуется. Это свидетельствует
о том, что давление с внешней стороны пузыря превышает давление внутри пузыря.
Иными словами, в состоянии равновесия давления в фазах, разделенных
искривленной поверхностью, различаются. Эта разность давлений называют капиллярным
давлением.
В большинстве случаев мы имеем дело с фазами, находящимися в равновесии с атмосферой. В этом случае давление воздуха можно рассматривать как постоянное, а капиллярное давление – как избыточное давление в конденсированной фазе, которое создается за счет искривления поверхности раздела фаз.
Количественная характеристика разности давлений по обе стороны искривленной поверхности выражается законом Лапласа – основным законом в теории капиллярности
, где – кривизна поверхности.
Если центр
кривизны лежит внутри жидкости, то кривизна положительна, если вне жидкости – отрицательна.
Из уравнения видно, что в фазе, имеющей положительную кривизну межфазной поверхности, давление больше, чем внутри фазы с отрицательной кривизной. Капиллярное давление всегда направлено к центру кривизны! Например, в столбике ртути, не смачивающей стеклянный капилляр (положительная кривизна , т. к. центр кривизны внутри фазы), давление ртути больше, чем в насыщенном паре над жидкой ртутью в капилляре. Если в капилляре находится смачивающая его вода, то радиус кривизны жидкой фазы будет отрицательным , а давление в этой фазе меньше, чем в насыщенном паре.
В общем случае , где r1 и r2 – главные радиусы кривизны.
В простейшем случае для сферической поверхности (пузырек или капля жидкости в невесомости) главные радиусы кривизны одинаковы и постоянны вдоль всей поверхности: ;
в случае цилиндра: .
Из представленных
выражений видно, что чем меньше радиус частиц (чем выше дисперсность), тем
больше радиус кривизны и больше капиллярное давление.
Как уже отмечалось выше, жидкость в капилляре может быть выше или ниже уровня жидкости в сосуде, куда опущен капилляр. Это зависит от того, смачивается капилляр жидкостью или нет. В первом случае жидкость будет подниматься, т. к. радиус кривизны жидкости отрицательный и дополнительное давление направлено в сторону газообразной фазы (в центр кривизны). Во втором случае жидкость будет опускаться (радиус кривизны жидкости положительный, дополнительное давление направлено внутрь жидкости). Два описанных варианта капиллярного движения жидкости схематически представлены на рисунках а и б.
Очевидно, что движение жидкости будет происходить до тех пор, пока давление Лапласа не уравновесится гидростатическим давлением столба жидкости высотой h. Для состояния равновесия можно записать равенство:
,
где ρ – плотность жидкости; ρо – плотность газовой
фазы; g –
ускорение свободного падения; r –
радиус мениска.
Из рисунка в выразим радиус капилляра ro через радиус мениска r и угол смачивания θ: rо=r∙cos θ. Тогда высоту капиллярного поднятия (опускания) можно выразить формулой Жюрена:
.
По формуле Жюрена можно рассчитать высоту капиллярного поднятия или опускания жидкости в зависимости от степени смачиваемости жидкостью материала капилляра, его радиуса и плотностей жидкой и газообразной фаз. Из анализа формулы следует, что если жидкость не смачивает капилляр (краевой угол θ>90°, а cos θ<0), то величина h будет отрицательной (плотность любой жидкости больше плотности воздуха), т. е. жидкость в капилляре опускается ниже уровня жидкости в сосуде. При смачивании будет наблюдаться обратная зависимость.
Итак, искривление
поверхности вызывает повышение или понижение давления в фазе по сравнению с
плоской поверхностью фазы такого же химического состава. Очевидно, что это
приводит к изменению термодинамических параметров вещества, которые определяют
его физические свойства и реакционную способность. Понятие термодинамическая
реакционная способность вещества характеризует его способность изменять
химический или фазовый состав, т. е. вступать в химическую реакцию или
переходить в новую фазу (например, испаряться или конденсироваться,
растворяться).
Понятие термодинамическая
реакционная способность вещества характеризует его способность изменять
химический или фазовый состав, т. е. вступать в химическую реакцию или
переходить в новую фазу (например, испаряться или конденсироваться,
растворяться).
Термодинамическая реакционная способность определяется величиной химического потенциала или другой функцией состояния (например, энергии Гиббса, если система находится при постоянных давлении и температуре, энергии Гельмгольца при постоянных объеме и температуре и др.). Напомним, что производная любой термодинамической функции состояния по соответствующему для данной системы параметру будет равна химическому потенциалу.
Для процесса испарения вещества зависимость давления насыщенного пара над жидкостью от кривизны поверхности выражается уравнением Кельвина (Томсона):
,
где pr – давление насыщенного пара
над искривленной поверхностью; p∞ –
давление насыщенного пара над плоской поверхностью; Vм – молярный объем жидкости.
Данное уравнение может быть применено для установления условий равновесия жидкости и пара при наличии между ними искривленной поверхности (например, в случае жидкости в капилляре или жидкости в виде капли). Из анализа уравнения Кельвина можно сделать вывод о том, что при положительной кривизне жидкости (капля в невесомости или на поверхности твердого тела при отсутствии или неполной смачиваемости) над ней создается повышенное по сравнению с плоской поверхностью давление пара, т. е. испаряется больше жидкости. При отрицательной кривизне (жидкость, смачивающая капилляр) количество испарившейся жидкости в равновесии с ее паром будет меньше по сравнению с плоской поверхностью; иными словами, конденсация будет происходить при меньшем давлении паров. Такое явление известно под названием «капиллярная конденсация».
Наряду со смачиванием
поверхности твердого тела жидкостью и растеканием жидкости по поверхности
другой жидкости, понижение поверхностной энергии системы может достигаться за
счет адсорбции – самопроизвольного перераспределения компонентов между объемом фазы
и ее поверхностным слоем. Адсорбция является универсальным процессом, так как
она характерна для любых поверхностей раздела фаз и встречается практически
повсеместно. Чаще всего под адсорбцией подразумевают концентрирование вещества
на твердой или жидкой поверхности, которое происходит вследствие перехода этого
вещества из объема одной или нескольких контактирующих фаз на межфазную
поверхность. Поглощающее (адсорбирующее) вещество называют адсорбентом, а
поглощающееся (адсорбирующееся) – адсорбатом. Здесь мы встречаемся с
удивительным явлением, в котором интенсивные величины (концентрации) в
самопроизвольном процессе не выравниваются, как обычно, а наоборот расходятся,
и в состоянии равновесия концентрации веществ в объемных фазах и на межфазных
поверхностях не равны. Этот результат адсорбционного процесса был предсказан
Гиббсом и после подтвержден экспериментально.
Адсорбция является универсальным процессом, так как
она характерна для любых поверхностей раздела фаз и встречается практически
повсеместно. Чаще всего под адсорбцией подразумевают концентрирование вещества
на твердой или жидкой поверхности, которое происходит вследствие перехода этого
вещества из объема одной или нескольких контактирующих фаз на межфазную
поверхность. Поглощающее (адсорбирующее) вещество называют адсорбентом, а
поглощающееся (адсорбирующееся) – адсорбатом. Здесь мы встречаемся с
удивительным явлением, в котором интенсивные величины (концентрации) в
самопроизвольном процессе не выравниваются, как обычно, а наоборот расходятся,
и в состоянии равновесия концентрации веществ в объемных фазах и на межфазных
поверхностях не равны. Этот результат адсорбционного процесса был предсказан
Гиббсом и после подтвержден экспериментально.
Наиболее яркий
пример адсорбции – это поглощение активированным углем вредных для здоровья
примесей из питьевой воды. Здесь адсорбентом является активированный уголь, а
адсорбатом – вредные примеси. Другой пример – концентрирование на поверхности любого
твердого тела или жидкости молекул различных примесей, находящихся в воздухе. При
растворении мыла в воде наблюдается понижение поверхностного натяжения образующегося
раствора за счет перехода растворенных молекул на его поверхность из объемной
фазы, что также относится к явлению адсорбции.
Здесь адсорбентом является активированный уголь, а
адсорбатом – вредные примеси. Другой пример – концентрирование на поверхности любого
твердого тела или жидкости молекул различных примесей, находящихся в воздухе. При
растворении мыла в воде наблюдается понижение поверхностного натяжения образующегося
раствора за счет перехода растворенных молекул на его поверхность из объемной
фазы, что также относится к явлению адсорбции.
Источники информации:
1. И. Е. Стась, А.С. Фомин. Дисперсные системы в природе и технике. – Барнаул, 2005. – 217 с.
2. Евстратова К.И., Купина И.А., Малахова Е.Е. Физическая химия. – М.: Высшая школа, 1990. – 487с.
3. Савицкая Т.А., Котиков Д.А. Пособие для самостоятельной работы над лекционным курсом «Коллоидная химия» в вопросах, ответах и упражнениях. – Минск, 2006. – 86 с.
4.
Амирханова
Н. А., Беляева Л.С., Белоногов В.А. Задачник по химии. – Уфа: Изд-во УГАТУ,
2002. – 117 с.
А., Беляева Л.С., Белоногов В.А. Задачник по химии. – Уфа: Изд-во УГАТУ,
2002. – 117 с.
5. Бугреева Е.В., Евстратова К.И., Купина Н.А. Практикум по физической и коллоидной химии. – М.: Высш. шк., 1990. – 255 с.
6. Материалы сайта http://www.tdmu.edu.te.ua/
Лекции / ФОМ. Лекции / lection / lec_15
9
1.Поверхностное натяжение
Молекулы
во внутренних слоях вещества испытывают
в среднем одинаковое по всем направлениям
притяжение со стороны окружающих
молекул, молекулы же поверхностного
слоя (рис. 127) подвергаются неодинаковому
притяжению со стороны внутренних слоев
вещества и со стороны, граничащей с
поверхностным слоем среды. Так, на
поверхности раздела жидкость —
воздух молекулы жидкости, находящиеся
в поверхностном слое, испытывают большее
притяжение со стороны соседних молекул
внутренних слоев жидкости, чем со стороны
молекул газа. Поэтому свойства
поверхностных слоев вещества всегда
несколько отличаются от свойств его
внутренних частей. Поверхностные
свойства оказывают влияние и на другие
свойства вещества. Если величина
поверхности веществ сравнительно
невелика, то эти влияния проявляются
слабо. Но по мере увеличения поверхности,
происходящего вследствие повышения
степени дисперсности (степени
раздробленности) вещества или увеличения
его пористости, влияние поверхностных
свойств начинает проявляться все сильнее
и становится значительным в случае,
когда вещества обладают сильно развитой
поверхностью. При дроблении вещества
суммарная поверхность его частиц
увеличивается весьма значительно.
Поэтому свойства
поверхностных слоев вещества всегда
несколько отличаются от свойств его
внутренних частей. Поверхностные
свойства оказывают влияние и на другие
свойства вещества. Если величина
поверхности веществ сравнительно
невелика, то эти влияния проявляются
слабо. Но по мере увеличения поверхности,
происходящего вследствие повышения
степени дисперсности (степени
раздробленности) вещества или увеличения
его пористости, влияние поверхностных
свойств начинает проявляться все сильнее
и становится значительным в случае,
когда вещества обладают сильно развитой
поверхностью. При дроблении вещества
суммарная поверхность его частиц
увеличивается весьма значительно.
Рис. 127. Поверхностный слой жидкости.
В
той или иной степени особенности свойств
поверхностных слоев вещества проявляются
на любой поверхности раздела между
двумя фазами. В однокомпонентных системах
они наблюдаются поверхностях раздела
жидкость—пар, твердое тело—пар, слабое
на границе раздела твердое тело —
жидкость и, наконец, еще более слабо—на
границе раздела между различными
кристаллическими модификациями. В
однокомпонентных системах эти особенности
обусловлены в основном разной концентрацией
вещества в различных фазах и в
некоторых случаях более или менее
закономерной ориентацией молекул
в поверхностном слое. В системах же,
содержащих более одного компонента,
и состав поверхностных слоев отличается
от состава внутренних частей фаз.
В
однокомпонентных системах эти особенности
обусловлены в основном разной концентрацией
вещества в различных фазах и в
некоторых случаях более или менее
закономерной ориентацией молекул
в поверхностном слое. В системах же,
содержащих более одного компонента,
и состав поверхностных слоев отличается
от состава внутренних частей фаз.
Указанные
особенности условий существования
молекул поверхностного слоя приводят
к тому, что для увеличения поверхности
требуется затратить работу. Величина
этой работы, отнесенная к 1 см2 поверхности, получила название
поверхностного натяжения. Как известно,
если какое-нибудь количество жидкости
поместить в среду другой жидкости,
не смешивающейся с ней и одинаковой
с ней плотности (чтобы исключить
искажающее влияние силы тяжести), то
жидкость принимает форму шара под
действием сил поверхностного натяжения,
всегда вызывающих уменьшение поверхности.
Чем меньше количество взятой жидкости,
тем сильнее проявляется этот эффект,
потому что при уменьшении размеров шара
объем его, а следовательно, и масса
уменьшаются в большей степени, чем
поверхность. (Объем шара пропорционален
третьей степени радиуса, а поверхность—только
его второй степени.) Капли жидкости
достаточно малых размеров приобретают
шарообразную форму даже в среде газа
(например, капли дождя или тумана).
Поверхностное натяжение будем обозначать
через а; его выражают в дин/см или в
эрг/см2 (эти значения численно совпадают). Таким
образом, поверхностное натяжение можно
выражать как силу на единицу длины или
как энергию на единицу поверхности.
Поверхностное натяжение представляет
не полную энергию по поверхности, а
равно максимальной полезной работе,
затрачиваемой на образование единицы
поверхности, т. е. представляет собой
удельный (на 1 см2)
изобарный потенциал поверхности.
(Объем шара пропорционален
третьей степени радиуса, а поверхность—только
его второй степени.) Капли жидкости
достаточно малых размеров приобретают
шарообразную форму даже в среде газа
(например, капли дождя или тумана).
Поверхностное натяжение будем обозначать
через а; его выражают в дин/см или в
эрг/см2 (эти значения численно совпадают). Таким
образом, поверхностное натяжение можно
выражать как силу на единицу длины или
как энергию на единицу поверхности.
Поверхностное натяжение представляет
не полную энергию по поверхности, а
равно максимальной полезной работе,
затрачиваемой на образование единицы
поверхности, т. е. представляет собой
удельный (на 1 см2)
изобарный потенциал поверхности.
Поверхностное
натяжение различных жидкостей
неодинаково, так как оно зависит от их
мольного объема, полярности молекул,
способности молекул к образованию
водородной связи между собой и пр.
Следует обратить внимание на большую
величину поверхностного натяжения воды
по сравнению с поверхностным натяжением
других обычных жидкостей. Еще выше
поверхностное натяжение расплавленных
солей и металлов (см., например,
поверхностное натяжение ртути).
Еще выше
поверхностное натяжение расплавленных
солей и металлов (см., например,
поверхностное натяжение ртути).
Поверхностное
натяжение жидкостей оказывает влияние
на многие их свойства. Соответственно
существуют различные методы измерения
поверхностного натяжения: по определению
высоты поднятия жидкости в капиллярной
трубке, по определению веса капель
жидкости при медленном вытекании ее с
конца вертикальной капиллярной
трубки (сталагмометра), по определению
максимального давления пузырьков
газа при пробулькивании его через
жидкость и др. (см. курс физики). На
поверхностях раздела твердых тел как
с газом, так и с жидкостью, несомненно,
имеют место такие же особенности
состояния молекул, атомов или ионов,
как и на поверхности раздела между
жидкостью и паром, однако методы прямого
измерения поверхностного натяжения
или общей энергии поверхности твердых
тел еще не разработаны. Ориентировочно
эти величины можно оценивать в некоторых
случаях по косвенным данным (по изменению
температуры плавления, растворимости,
давления насыщенного пара и другим).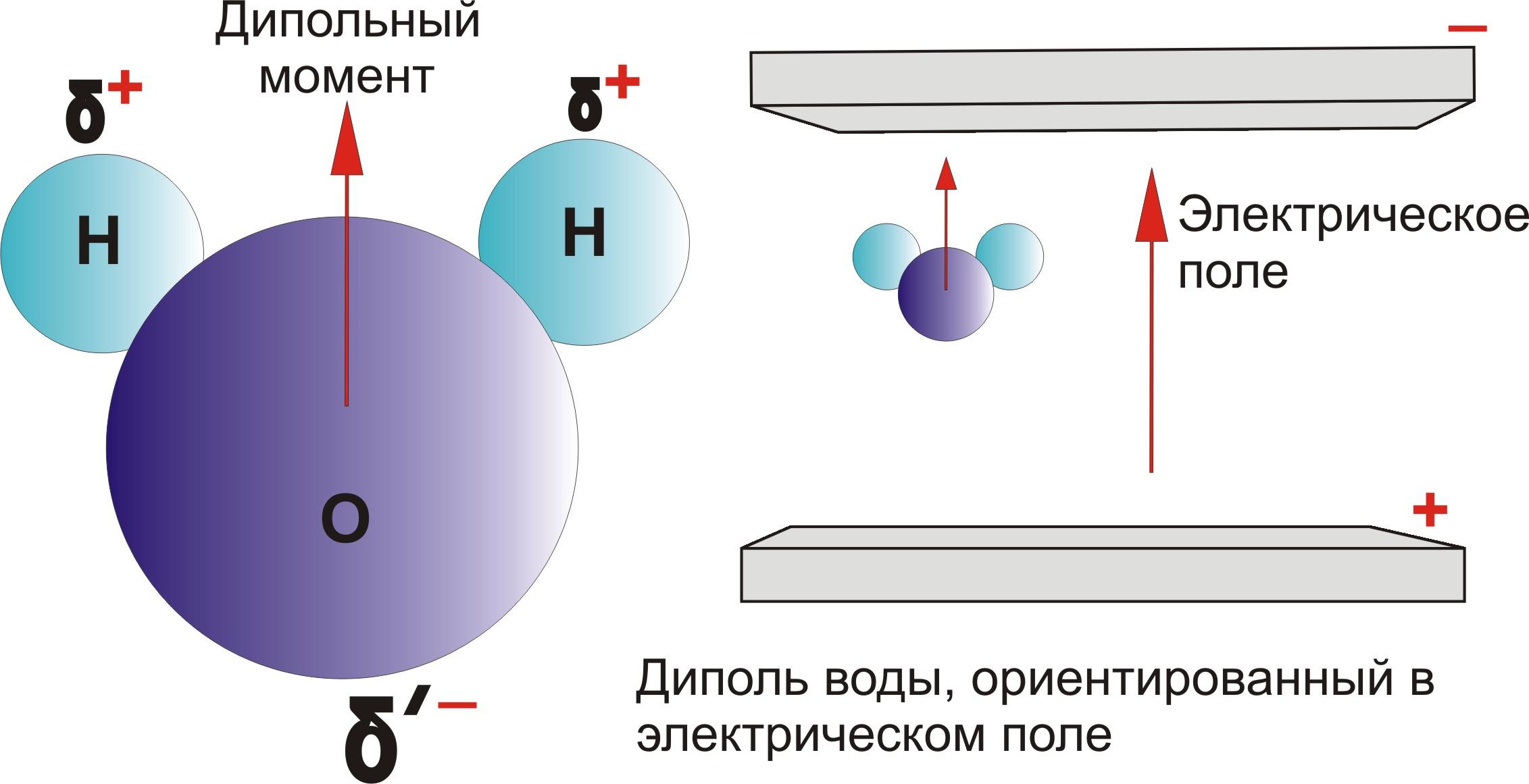 Такие определения являются еще очень
неточными и результаты их недостаточно
согласуются между собой.
Такие определения являются еще очень
неточными и результаты их недостаточно
согласуются между собой.
Рис. 128. Зависимость поверхностного натяжения некоторых жидкостей от температуры
При
повышении температуры вещество
расширяется, ослабляются силы
взаимного притяжения между молекулами
внутри вещества и в поверхностном
слое. Поэтому с повышением температуры
поверхностное натяжение уменьшается.
При температурах более высоких, чем
нормальная температура кипения данной
жидкости, поверхностное натяжение
измеряют уже не при атмосферном
давлении, а при давлении насыщенного
пара. Если результаты измерений
представить графически, отложив
поверхностное натяжение как функцию
температуры (рис. 128), то зависимость
для многих веществ оказывается линейной,
почти вплоть до критической температуры,
при которой поверхностное натяжение
становится равным нулю, так как
исчезает различие между жидкостью
и паром. Основываясь на линейном
уменьшении поверхностного натяжения
с повышением температуры, Менделеев
установил (1860) существование такой
температуры, при которой поверхностное
натяжение становится равным нулю. Выше этой температуры вещество уже не
может находиться в жидком состоянии.
Эту температуру Менделеев назвал
температурой абсолютного кипения
(позднее ее стали называть критической
температурой).
Выше этой температуры вещество уже не
может находиться в жидком состоянии.
Эту температуру Менделеев назвал
температурой абсолютного кипения
(позднее ее стали называть критической
температурой).
2.Адсорбция Анализ гетерогенных равновесии
показывает, что непременным условием
их существования является наличие
границы раздела фаз. Состояние атомов
или молекул на границе отличается от
состояния в объеме фаз вследствие
нескомпенсированности атомных полей
частиц, выходящих на поверхность.
Состояние поверхности и поверхностные
силы играют существенную роль в тех
случаях, когда поверхность сильно
развита, например, при раздробленном
мелкодисперсном состоянии вещества
либо при получении его в виде тонких
пленок, когда сфера действия приповерхностных
сил соизмерима с толщиной пленок. Следует
отметить, что при анализе гетерогенных
равновесии предполагается, что каждая
фаза во всех ее точках совершенно
однородна, т. е. состав ее всюду одинаков.
В действительности вблизи поверхности
раздела двух фаз это условие не
соблюдается, и концентрации компонентов
отличаются от концентраций в объемах
сосуществующих фаз. Например,
концентрация газа у поверхности
какой-нибудь твердой или жидкой фазы
возрастает—положительная адсорбция.
Обратный случай—уменьшение концентрации
какого-либо компонента вблизи поверхности
раздела—называют отрицательной
адсорбцией.
Например,
концентрация газа у поверхности
какой-нибудь твердой или жидкой фазы
возрастает—положительная адсорбция.
Обратный случай—уменьшение концентрации
какого-либо компонента вблизи поверхности
раздела—называют отрицательной
адсорбцией.
Количество вещества, поглощенное или адсорбированное единицей поверхности раздела, называют поверхностной концентрацией или поверхностной плотностью данного вещества и обозначают буквой Г. Эта величина является еще одной переменной, новой по отношению к рассматривавшимся ранее при анализе гетерогенных равновесий. Вещество, на поверхности которого происходит адсорбция, называется адсорбентом, а поглощаемое из соседней или окружающей фазы — абсорбатом. Явление адсорбции впервые наблюдал шведский ученый Карл Шееле (1773) в Швеции.
Адсорбционные
явления на поверхности твердого тела
относят к физической адсорбции, если
молекулы адсорбата сохраняют
свою
индивидуальность, а силы адсорбции
аналогичны силам Ван-дер-Ваальса в
реальных газах, или к химической
адсорбции, если адсорбируемая молекула
образует с твердым телом химическое
соединение, что сопровождается обменным
взаимодействием, включающим и межионное
взаимодействие. Согласно Леннард-Джонсу,
потенциальная кривая (рис. 66) характеризует
химическую адсорбцию, а кривая 2—физическую
адсорбцию.
Согласно Леннард-Джонсу,
потенциальная кривая (рис. 66) характеризует
химическую адсорбцию, а кривая 2—физическую
адсорбцию.
Рис. 66. Потенциальные кривые взаимодействия при химической (1) и физической (2) адсорбции
Видно,
что кривая 1 обладает более глубоким
минимумом. Равновесные расстояния г1 и г2 определяют положение минимума
потенциальной энергии. Для физической
адсорбции г1 всегда больше г2 для
химической. Потенциальный барьер, высота
которого отождествляется с энергией
активации, соответствует точке
пересечения кривых 1 и 2. Теоретически,
рассматривая физическую адсорбцию,
полагают, что твердое тело и адсорбат
являются двумя независимыми системами.
Воздействие адсорбируемой молекулы на
решетку твердого тела принимается за
слабое возмущение. В случае химической
адсорбции адсорбент и адсорбат
рассматриваются как единая
квантомеханическая система. Разные
пути подхода к рассмотрению
адсорбционной системы требуют надежных
критериев, дающих возможность различать
оба типа адсорбции.
3.Адсорбционная формула Гиббса.
Полная поверхностная энергия слоя может быть записана
Для полного дифференцила на основании (1)
dU =
Это выражение совместимо с уравнением
(ХП1.87)
при условии (4)
Следовательно, при постоянной температуре согласно (4) имеем
(5)
Уравнения (1) и (4) получены Гиббсом и являются фундаментальными термодинамическими соотношениями для межфазного поверхностного слоя. Поскольку величины, входящие в уравнения (1) и (4), зависят от значения поверхности раздела, то целесообразно рассматривать абсолютные величины, отнесенные к единице поверхности:
Величина
Гi,
введенная Гиббсом, есть избыток числа
молей
i-гo компонента
в объеме поверхностного слоя площадью
s=l
по сравнению с числом молей в том же
объеме, если бы смежные фазы переходили
одна в другую без изменения плотности. Эта величина называется абсолютной
величиной адсорбции 1-го компонента у
данной поверхности. Для определения
направленности адсорбционных
процессов и устойчивости образующихся
слоев целесообразно определить
избыточную свободную энергию Гельмгольца
поверхностного слоя или свободную
поверхностную энергию Гельмгольца.
В соответствии с уравнением (1) можно
записать
Эта величина называется абсолютной
величиной адсорбции 1-го компонента у
данной поверхности. Для определения
направленности адсорбционных
процессов и устойчивости образующихся
слоев целесообразно определить
избыточную свободную энергию Гельмгольца
поверхностного слоя или свободную
поверхностную энергию Гельмгольца.
В соответствии с уравнением (1) можно
записать
Из этого уравнения при учете (4) имеем
При постоянстве Т и ni на основании (XIII.115) имеем
Отсюда можно заключить, что межфазное поверхностное натяжение представляет собой свободную поверхностную энергию Гельмгольца единицы поверхности при постоянных Т, V и постоянной концентрации слоя. Иначе поверхностное натяжение можно рассматривать как силу, действующую на единицу длины контура поверхности раздела фаз. Из уравнений (5) и ( .112) следует, что при постоянной температуре для единицы поверхности изменение поверхностного натяжения определяется следующим выражением:
(XIII. 117)
117)
Это уравнение и есть известная адсорбционная формула Гиббса. Свободная поверхностная энергия Гельмгольца слоя с площадью, равной единице, согласно (XIII.114) определится выражением (XIII. 118)
Если рассматривать систему из двух практически нерастворимых друг в друге компонентов, один из которых образует жидкую, а другой газовую фазу, то адсорбционное уравнение Гиббса (XIII.117) принимает следующий вид:
.119)
Поскольку влиянием растворимости можно пренебречь, химический потенциал компонента, образующего жидкую фазу, практически не изменяется (d =0) и, следовательно, из уравнения (XIII.119) получаем
(.120)
Учитывая, что изменение химического потенциала идеального газа при давлении р определяется из уравнения (VI.51), на основании (XIII.120) получаем
(XIII.121)
Таким
образом, определяя зависимость
поверхностного натяжения в нерастворяющей
жидкости (компонента) от давления пара
второго компонента, получаем величину
адсорбции этого компонента на поверхности
жидкости. Схематически типовая зависимость
поверхностного натяжения от
парциального давления и рассчитанная
на ее основе с помощью уравнения (
.121)
кривая адсорбции представлены на
рис. 71.
Схематически типовая зависимость
поверхностного натяжения от
парциального давления и рассчитанная
на ее основе с помощью уравнения (
.121)
кривая адсорбции представлены на
рис. 71.
Если второй компонент растворяется в объеме первого, то, очевидно, химический потенциал растворителя будет изменяться. Однако, выбирая положение поверхности, относительно которой определяется величина адсорбции, таким образом, чтобы адсорбция самого растворителя оказалась равной нулю (Г1==0), можно использовать уравнение ( .120). Указанного положения можно достичь, перемещая поверхность в сторону фазы (‘) или фазы (//) до тех пор, пока положительный избыток первого компонента по одну сторону поверхности не будет точно равен отрицательному избытку его по другую сторону. В этом случае
(XIII.122)
Индекс
(/)
у величины адсорбции Г2 указывает на выбор положения
поверхности, при котором
Г1=0. Такой прием является искусственным
и представляет известные неудобства
вследствие неопределенности выбора
указанного положения, однако при малых
концентрациях С2 объемного раствора в случае достаточно
сильно адсорбирующихся веществ
положение поверхности, при котором
Г1 =0,
практически не отличается от физической
поверхности раздела между фазами.
Такой прием является искусственным
и представляет известные неудобства
вследствие неопределенности выбора
указанного положения, однако при малых
концентрациях С2 объемного раствора в случае достаточно
сильно адсорбирующихся веществ
положение поверхности, при котором
Г1 =0,
практически не отличается от физической
поверхности раздела между фазами.
Принимая во внимание уравнение (VI.63) в его дифференциальной форме, на основании (XIII.121) будем иметь
Учитывая, что в сильно разбавленных растворах , получим
С помощью этой формулы можно рассчитать изотерму адсорбции, зная концентрационную зависимость поверхностного натяжения.
Предельное значение понижения поверхностного натяжения с концентрацией, т. е. величина
называется
поверхностной активностью. Те вещества,
которые при добавлении к данному
растворителю снижают его поверхностное
натяжение,. называются поверхностно-активными.
Для поверхностно-активных веществ
называются поверхностно-активными.
Для поверхностно-активных веществ
(XIII. 125)
Вещества, повышающие поверхностное натяжение, адсорбируются отрицательно. Такие вещества называются поверхностно-инактивными. Для поверхностно-инактивных веществ
Рис. 71. Зависимость поверхностного натяжения от парциального давления (а) и рассчитанная по уравнению (XIII.121) кривая адсорбции (б) | Рис. 72. Изотермы поверхностного натяжения (а) и адсорбции (б): /—для поверхностно-активных; 2— для поверхностно-инактивных веществ |
Следует
подчеркнуть, что в случае
поверхностно-инактивных веществ
концентрация растворителя в поверхностном
слое изменяется слабо и величина
отрицательной адсорбции, следовательно,
незначительна. На рис. 72 схематически
представлена концентрационная
зависимость поверхностного натяжения
и рассчитанные на ее основе при помощи
уравнения (XIII. 124) кривые адсорбции
соответственно для поверхностно-активных
(кривая 1) и поверхностно-инактивных
(кривая 2) веществ.
На рис. 72 схематически
представлена концентрационная
зависимость поверхностного натяжения
и рассчитанные на ее основе при помощи
уравнения (XIII. 124) кривые адсорбции
соответственно для поверхностно-активных
(кривая 1) и поверхностно-инактивных
(кривая 2) веществ.
§ 151. Практическое применение адсорбции. Адсорбция находит разностороннее применение. Мы уже упоминали о том, что при гетерогенном катализе как в газовой среде, так и в растворах процесс адсорбции реагирующих веществ твердым катализатором обычно играет решающую роль. Широко применяются твердые адсорбенты также и в различных процессах очистки газов или растворов от нежелательных примесей или загрязнений. Сюда относится, в частности, применение активированного угля для противогазов, введение благодаря работам Н. Д. Зелинского, спасшего этим много тысяч человеческих жизней. Сюда же относятся и многие процессы очистки и осушки различных газов в пронзвод-С1——ыя условиях и, наконец, процессы осветлеяня и обесцвечива
ния
растворов в производствах сахара,
глюкозы, нефтепродуктов, некоторых
фармацевтических препаратов и др.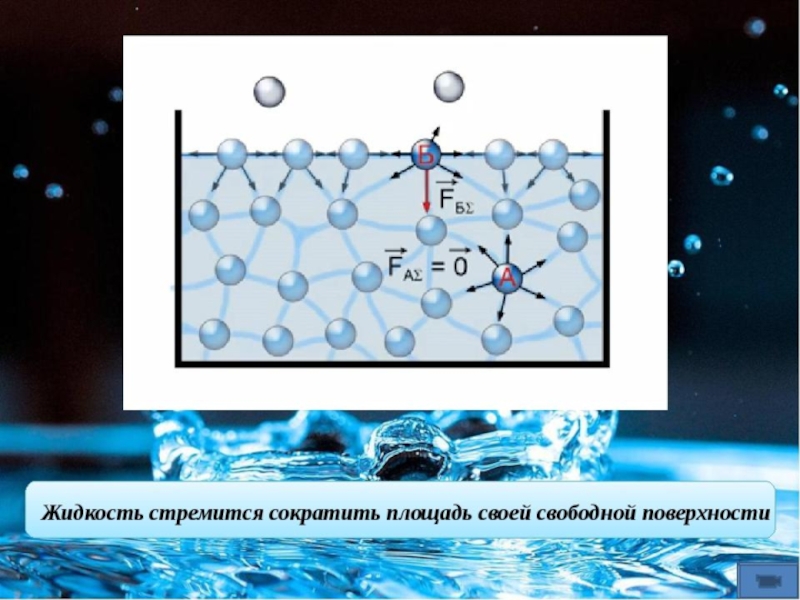 Иногда
процесс адсорбции применяется для
извлечения какого< нибудь ценного
продукта, находящегося в виде примеси
в газе или в растворах, например в
процессах рекуперации летучих раствори<
телей воздух, содержащий пары ценного
растворителя (бензола, ацетона и др.),
пропускают через слой активного угля
или силика-геля, который их адсорбирует.
Путем последующего нагревания адсорбента
или продувкой его водяным паром
растворители можно выделить в чистом
виде. Большую роль адсорбционные явления
играют и в процессах крашения. Так, при
крашении шерсти обычно происходит
сначала адсорбция красителя, за которой
следует уже химическая реакция в
адсорбционном слое. Свойства многих
порошкообразных материалов, в частности
соответствующих строительных материалов,
могут существенно изменяться при
адсорбции на их поверхности тех или
других веществ. На этом основана,
например, гидрофобизация цемента при
обработке его растворами солей
высокомолекулярных органических кислот
и др. Почвой адсорбируются различные
растворенные вещества из природных
вод.
Иногда
процесс адсорбции применяется для
извлечения какого< нибудь ценного
продукта, находящегося в виде примеси
в газе или в растворах, например в
процессах рекуперации летучих раствори<
телей воздух, содержащий пары ценного
растворителя (бензола, ацетона и др.),
пропускают через слой активного угля
или силика-геля, который их адсорбирует.
Путем последующего нагревания адсорбента
или продувкой его водяным паром
растворители можно выделить в чистом
виде. Большую роль адсорбционные явления
играют и в процессах крашения. Так, при
крашении шерсти обычно происходит
сначала адсорбция красителя, за которой
следует уже химическая реакция в
адсорбционном слое. Свойства многих
порошкообразных материалов, в частности
соответствующих строительных материалов,
могут существенно изменяться при
адсорбции на их поверхности тех или
других веществ. На этом основана,
например, гидрофобизация цемента при
обработке его растворами солей
высокомолекулярных органических кислот
и др. Почвой адсорбируются различные
растворенные вещества из природных
вод. П. А. Ребиндер нашел, что адсорбционные
процессы могут приводить к понижению
прочности некоторых материалов (металлов,
горных пород) и это дает возможность
интенсифицировать процессы их механической
обработки. Коллоидные системы
вследствие очень малых размеров частиц
обладают настолько большой поверхностью
раздела, что адсорбционные процессы
развиваются на них особенно интенсивно.
П. А. Ребиндер нашел, что адсорбционные
процессы могут приводить к понижению
прочности некоторых материалов (металлов,
горных пород) и это дает возможность
интенсифицировать процессы их механической
обработки. Коллоидные системы
вследствие очень малых размеров частиц
обладают настолько большой поверхностью
раздела, что адсорбционные процессы
развиваются на них особенно интенсивно.
ЛЕКЦИЯ 16
Поверхностные пленки на твердых телах.
Адсорбция газов на поверхности твердых тел охватывает как явления чисто физической адсорбции, близкой к процессам физической конденсации пара в жидкость, так и явления химической адсорбции.
Остановимся
теперь вкратце на явлении, которое можно
рассматривать как дальнейшее развитие
этих последних процессов. Давно уже
известно, что некоторые металлы, например
алюминий, магний, свинец, в атмосферных
условиях, взаимодействуя с кислородом
воздуха, окисляются с поверхности и
покрываются тонкой пленкой окиси,
которая благодаря своей компактности
изолирует внутренние слои металла
от соприкосновения с воздухом и этим
защищает металл от дальнейшего окисления. Образование окисной пленки на поверхности
свойственно почти всем металлам, включая
сюда медь, никель, хром и другие металлы,
считавшиеся долгое время вполне
устойчивыми к таким воздействиям. Однако
на этих металлах толщина образующихся
пленок во много раз меньше толщины тех
пленок, существование которых было
установлено ранее. Эти более тонкие
пленки не изменяют внешнего вида
поверхности металла и не обнаруживаются
глазом.
Образование окисной пленки на поверхности
свойственно почти всем металлам, включая
сюда медь, никель, хром и другие металлы,
считавшиеся долгое время вполне
устойчивыми к таким воздействиям. Однако
на этих металлах толщина образующихся
пленок во много раз меньше толщины тех
пленок, существование которых было
установлено ранее. Эти более тонкие
пленки не изменяют внешнего вида
поверхности металла и не обнаруживаются
глазом.
Рис. 138. Рост окислительной пленки на меди при различных температурах.
На
рис. 138 изображены кривые роста окисной
пленки на меди при различных температурах.
Они показывают, что толщина пленки
сильно возрастает с повышением
температуры. Исследование процессов
образования таких пленок на различных
металлах показало, что толщина их в
зависимости от условий колеблется в
пределах от 10—15 до 200—400 А. Начиная с
толщины 200—400 А, пленки уже изменяют
внешний вид металла и при дальнейшем
увеличении толщины придают ему цвет,
свойственный данному окислу. Очевидно,
что пленки состоят не из одного слоя
молекул окисла, а содержат различное и
иногда значительное число их. Нужно
заметить, что свойства вещества в такой
поверхностной пленке могут в той или
иной степени отличаться от свойств его
в свободном состоянии.
Очевидно,
что пленки состоят не из одного слоя
молекул окисла, а содержат различное и
иногда значительное число их. Нужно
заметить, что свойства вещества в такой
поверхностной пленке могут в той или
иной степени отличаться от свойств его
в свободном состоянии.
Первоначальное предположение, что рост пленки всегда происходит в результате диффузии атомов кислорода в глубь металла, не подтвердилось при более глубоком изучении механизма этого процесса. Оказалось, что в ряде систем рост пленки обусловливается диффузией атомов (или, правильнее, ионов) металла к внешней поверхности образующейся пленки. Образование окисных пленок происходит не только под действием кислорода воздуха, но и при воздействии различными окислителями в растворах и путем окисления электрическим током (анодного окисления).
Пленки
на поверхности металлов могут быть не
только окисные. Например, при действии
на серебро галогенами (газообразными
или растворенными в органических
растворителях) поверхность его покрывается
пленкой соответствующего галогенида
серебра; свинец под действием серной
кислоты покрывается пленкой нерастворимого
сульфата свинца, защищающего металл от
дальнейшего взаимодействия.
Иногда металл защищают путем покрытия его поверхности фосфатными пленками и др. Подобные пленки могут образовываться и не только на металлах. Так, окись магния при взаимодействии с водой гидратируется и покрывается с поверхности пленкой гидрата окиси, нерастворимой в воде и изолирующей окись от дальнейшего воздействия.
В
каких же случаях поверхностные пленки
являются устойчивыми, удерживаются
на поверхности, образуя плотный слой,
и обладают в соответствии с этим защитными
свойствами? Несомненно, что здесь
играют роль многие факторы, и в разных
системах из них могут быть преобладающими.
Работами Д. Данкова было установлено,
что по крайней мере для окисных пленок
на металлах основным фактором служит кристаллохимическое
соответствие структур металлов и пленки. Так, на железе пленка,
состоящая из кристаллов окиси железа
кубической структуры (у-Fе2 Оз),
благодаря соответствию этой структуры
структуре металла может удерживаться
на поверхности, образуя компактный
слой и проявляя защитные свойства.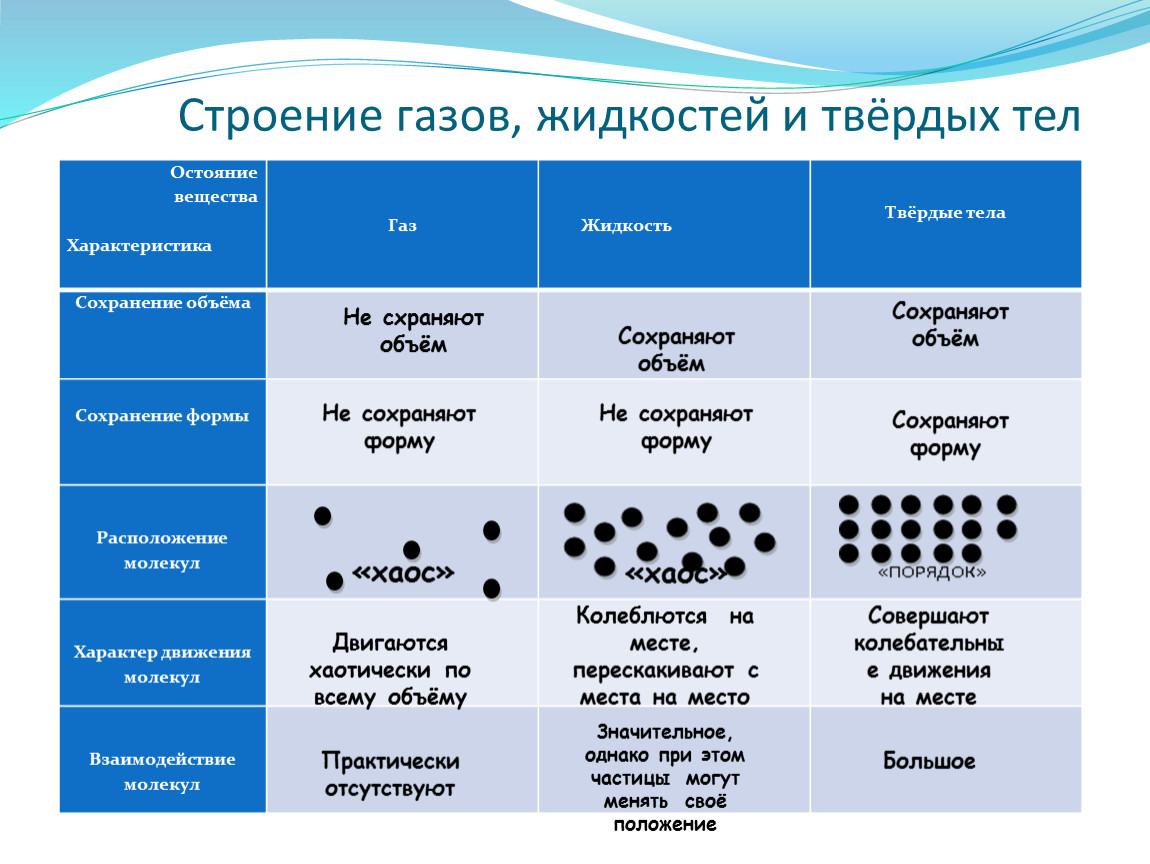 При
нагревании же до высокой температуры,
вследствие перехода окиси железа в
другую модификацию (а-РегОз), защитные
свойства пленки исчезают. По той же
причине ржавчина, представляющая собой
гидратированную окись железа (точнее,
y-Fe2O3-H2O),
образующуюся во влажном воздухе и
обладающую ромбической структурой
(отличной от железа), не проявляет
защитных свойств.
При
нагревании же до высокой температуры,
вследствие перехода окиси железа в
другую модификацию (а-РегОз), защитные
свойства пленки исчезают. По той же
причине ржавчина, представляющая собой
гидратированную окись железа (точнее,
y-Fe2O3-H2O),
образующуюся во влажном воздухе и
обладающую ромбической структурой
(отличной от железа), не проявляет
защитных свойств.
В
заключение остановимся на другом важном
явлении — на влиянии, которое твердое
тело оказывает на прилегающие к нему
слои жидкости. Раньше считали, что такое
влияние ограничивается лишь прилегающим
к поверхности мономолекулярным слоем.
Этот взгляд был опровергнут исследованиями
Б. В. Дерягина, показавшими, что
особенности в свойствах обнаруживаются
в слоях жидкости толщиной в десятки
и сотни молекул. При изучении свойств
этих слоев было обнаружено, что слои
жидкости, прилегающие к поверхности
твердого тела, могут изменять свойства
на расстоянии 10 —6 и даже 10-5 см от поверхности.
Особенно рельефно это обнаруживается при изучении свойств воды, образующейся при конденсации из ненасыщенного пара в узких капиллярах из силикатного или кварцевого стекла. Получающиеся таким путем тонкие слои воды могут обладать более плотной структурой, повышенной вязкостью и в определенных условиях даже довольно значительным модулем сдвига. Для них наблюдается изменение коэффициента термического расширения и соответственно изменение температуры, отвечающей максимальной плотности. Такие структуры обладают значительной стойкостью во времени и выдерживают без разрушения нагревание (в запаянных капиллярах). Превращение их в лед может потребовать переохлаждения до -40 или -50 °С.
Экспериментальные
данные, полученные в других условиях,
подтверждают, что слон жидкости,
прилегающие к поверхности твердого
тела, могут обладать отличительными
свойствами и, следовательно, иным
строением. В той или другой форме это
наблюдалось для разных жидкостей. Остановимся лишь на воде, так как для
нее это явление довольно хорошо изучено
экспериментально. Рассмотренные ранее
процессы взаимодействия молекул воды
с ионами и атомами в кристаллогидратах
показывают, что эти молекулы могут
подобным же образом взаимодействовать
и с ионами или атомами, содержащимися
в поверхностном слое кристалла или
стекла. Взаимодействие может приводить
к образованию более или менее прочной
донорно-акцепторной связи и водородной
связи или ионо-днпольной связи, причем
наряду с типичными случаями здесь
возможны и переходные формы взаимодействия,
когда деление соединений по характеру
связи становится условным. Такое
взаимодействие, связывая молекулу воды
с поверхностью кристалла, вызывает
преимущественную ориентацию ее
относительно поверхности, способствуя
образованию упорядоченного расположения
молекул относительно поверхности.
Рассмотренное взаимодействие может
вместе с тем вызывать дополнительную
поляризацию молекул воды, что повышает
их способность связывать другие молекулы
воды, расположенные дальше от поверхности,
увеличивая полярность этих молекул, но
уже в меньшей степени.
Остановимся лишь на воде, так как для
нее это явление довольно хорошо изучено
экспериментально. Рассмотренные ранее
процессы взаимодействия молекул воды
с ионами и атомами в кристаллогидратах
показывают, что эти молекулы могут
подобным же образом взаимодействовать
и с ионами или атомами, содержащимися
в поверхностном слое кристалла или
стекла. Взаимодействие может приводить
к образованию более или менее прочной
донорно-акцепторной связи и водородной
связи или ионо-днпольной связи, причем
наряду с типичными случаями здесь
возможны и переходные формы взаимодействия,
когда деление соединений по характеру
связи становится условным. Такое
взаимодействие, связывая молекулу воды
с поверхностью кристалла, вызывает
преимущественную ориентацию ее
относительно поверхности, способствуя
образованию упорядоченного расположения
молекул относительно поверхности.
Рассмотренное взаимодействие может
вместе с тем вызывать дополнительную
поляризацию молекул воды, что повышает
их способность связывать другие молекулы
воды, расположенные дальше от поверхности,
увеличивая полярность этих молекул, но
уже в меньшей степени. Это в свою очередь
усиливает связь с ними следующих молекул
воды, но еще в меньшей степени.
Это в свою очередь
усиливает связь с ними следующих молекул
воды, но еще в меньшей степени.
Вследствие
искажения направления связей в
дополнительно поляризованных
молекулах при упорядоченном расположении
их под ориентирующим действием поверхности
кристалла энергетически более
преимущественными могут быть структуры,
отличные от структур, свойственных
обычной воде и обычному льду. На это
указывает более высокая плотность
структуры рассматриваемых слоев воды.
Особенности внутреннего строения и
свойств тонких слоев воды, прилегающих
к твердой поверхности, и воды, находящейся
в тонких капиллярах, представляет
большой интерес для понимания свойств
многих биологических систем, слоистых
минералов, слоистых и дисперсных
горных пород, коллоидных систем и др.
Так, понижение температуры до 0°С (и
несколько ниже) может не вызывать
перехода воды тонких слоев в обычный
лед. Во-первых, потому что в условиях
влияния поверхности большая устойчивость
структуры льда по сравнению со структурой
тонкого слоя может достигаться не при
0°С, а при более низких температурах. Во-вторых, потому, что такое изменение
структуры требует разрыва существующих
связей. Рассматривая подобные процессы,
нельзя упускать из вида релаксационный
характер их и сильное уменьшение скорости
релаксации с понижением температуры.
В природных процессах все соотношения
дополнительно усложняются еще тем,
что вместо чистой
H2O
в них участвует природная вода, содержащая
различные
растворенные вещества, в том числе соли
и другие
Во-вторых, потому, что такое изменение
структуры требует разрыва существующих
связей. Рассматривая подобные процессы,
нельзя упускать из вида релаксационный
характер их и сильное уменьшение скорости
релаксации с понижением температуры.
В природных процессах все соотношения
дополнительно усложняются еще тем,
что вместо чистой
H2O
в них участвует природная вода, содержащая
различные
растворенные вещества, в том числе соли
и другие
фазовых диаграмм | Химия для специальностей
Результаты обучения
- Объяснить построение и использование типичной фазовой диаграммы
- Использование фазовых диаграмм для идентификации стабильных фаз при заданных температурах и давлениях, а также для описания фазовых переходов, происходящих в результате изменения этих свойств
- Описать сверхкритическую флюидную фазу вещества
В предыдущем модуле было описано изменение равновесного давления паров жидкости в зависимости от температуры. Принимая во внимание определение точки кипения, графики зависимости давления пара от температуры показывают, как точка кипения жидкости зависит от давления. Описано также использование кривых нагрева и охлаждения для определения температуры плавления (или замерзания) вещества. Выполнение таких измерений в широком диапазоне давлений дает данные, которые можно представить графически в виде фазовой диаграммы. А 9Фазовая диаграмма 0013 объединяет графики зависимости давления от температуры для равновесий фазовых переходов жидкость-газ, твердое тело-жидкость и твердое тело-газ. На этих диаграммах показаны физические состояния, существующие при определенных условиях давления и температуры, а также приведена зависимость от давления температур фазовых переходов (температуры плавления, сублимации, кипения). Типичная фазовая диаграмма чистого вещества показана на рис. 1.
Описано также использование кривых нагрева и охлаждения для определения температуры плавления (или замерзания) вещества. Выполнение таких измерений в широком диапазоне давлений дает данные, которые можно представить графически в виде фазовой диаграммы. А 9Фазовая диаграмма 0013 объединяет графики зависимости давления от температуры для равновесий фазовых переходов жидкость-газ, твердое тело-жидкость и твердое тело-газ. На этих диаграммах показаны физические состояния, существующие при определенных условиях давления и температуры, а также приведена зависимость от давления температур фазовых переходов (температуры плавления, сублимации, кипения). Типичная фазовая диаграмма чистого вещества показана на рис. 1.
Рис. 1. Физическое состояние вещества и температуры его фазовых переходов представлены графически на фазовой диаграмме.
Чтобы проиллюстрировать полезность этих графиков, рассмотрим фазовую диаграмму воды, показанную на рис. 2.
важные свойства.
Мы можем использовать фазовую диаграмму для определения физического состояния образца воды при заданных условиях давления и температуры. Например, давление 50 кПа и температура -10 °С соответствуют области диаграммы, обозначенной как «лед». В этих условиях вода существует только в твердом состоянии (лед). Давление 50 кПа и температура 50 °С соответствуют области «вода» — здесь вода существует только в виде жидкости. При 25 кПа и 200 °C вода существует только в газообразном состоянии. Обратите внимание, что на H 2 O фазовая диаграмма, оси давления и температуры не вычерчены в постоянной шкале, чтобы можно было проиллюстрировать несколько важных особенностей, как описано здесь.
Кривая BC на рисунке 2 представляет собой график зависимости давления паров от температуры, как описано в предыдущем модуле этой главы. Эта кривая «жидкость-пар» разделяет жидкую и газообразную области фазовой диаграммы и дает точку кипения воды при любом давлении. Например, при 1 атм температура кипения составляет 100°С. Обратите внимание, что кривая жидкость-пар заканчивается при температуре 374 °C и давлении 218 атм, что указывает на то, что вода не может существовать в виде жидкости выше этой температуры, независимо от давления. Физические свойства воды в этих условиях занимают промежуточное положение между свойствами ее жидкой и газообразной фаз. Это уникальное состояние вещества называется сверхкритической жидкостью, и эта тема будет описана в следующем разделе этого модуля.
Обратите внимание, что кривая жидкость-пар заканчивается при температуре 374 °C и давлении 218 атм, что указывает на то, что вода не может существовать в виде жидкости выше этой температуры, независимо от давления. Физические свойства воды в этих условиях занимают промежуточное положение между свойствами ее жидкой и газообразной фаз. Это уникальное состояние вещества называется сверхкритической жидкостью, и эта тема будет описана в следующем разделе этого модуля.
Рисунок 3 Сублимированные продукты, такие как это мороженое, обезвоживаются сублимацией при давлении ниже тройной точки для воды. (кредит: ʺlwaoʺ/Flickr)
Кривая твердого тела и пара, обозначенная AB на рис. 2, показывает температуры и давления, при которых лед и водяной пар находятся в равновесии. Эти пары данных температура-давление соответствуют точкам сублимации или осаждения воды. Если бы мы могли увеличить линию твердого газа на рисунке 2, мы бы увидели, что лед имеет давление паров около 0,20 кПа при температуре -10 °C. Так, если поместить замороженный образец в вакуум с давлением менее 0,20 кПа, лед возгонится. Это лежит в основе процесса «сублимационной сушки», часто используемого для сохранения пищевых продуктов, таких как мороженое, показанное на рис. 3.
Так, если поместить замороженный образец в вакуум с давлением менее 0,20 кПа, лед возгонится. Это лежит в основе процесса «сублимационной сушки», часто используемого для сохранения пищевых продуктов, таких как мороженое, показанное на рис. 3.
Кривая твердое тело-жидкость, обозначенная BD, показывает температуры и давления, при которых лед и жидкая вода находятся в равновесии, представляя точки плавления/замерзания воды. Обратите внимание, что эта кривая имеет небольшой отрицательный наклон (сильно преувеличенный для ясности), что указывает на то, что температура плавления воды немного снижается при увеличении давления. В этом отношении вода является необычным веществом, так как у большинства веществ температура плавления повышается с повышением давления. Такое поведение частично отвечает за движение ледников, подобное тому, что показано на рис. 4. Дно ледника испытывает огромное давление из-за своего веса, которое может растопить часть льда, образуя слой жидкой воды, на котором скользит ледник. может легче скользить.
может легче скользить.
Рис. 4. Огромное давление под ледниками приводит к их частичному таянию, образуя слой воды, обеспечивающий смазку, способствующую движению ледников. На этом спутниковом снимке показан наступающий край ледника Перито-Морено в Аргентине. (кредит: НАСА)
Точка пересечения всех трех кривых обозначена буквой B на рисунке 2. При давлении и температуре, представленных этой точкой, все три фазы воды сосуществуют в равновесии. Эта пара данных температура-давление называется тройная точка . При давлении ниже тройной точки вода не может существовать в жидком состоянии независимо от температуры.
Пример 1:
Определение состояния водыИспользуя фазовую диаграмму для воды, приведенную на рисунке 10.30, определите состояние воды при следующих температурах и давлениях:
- -10 °C и 50 кПа
- 25 °C и 90 кПа
- 50 °C и 40 кПа
- 80 °C и 5 кПа
- −10 °C и 0,3 кПа
- 50 °C и 0,3 кПа
Какие фазовые изменения может претерпевать вода при изменении температуры, если давление поддерживается на уровне 0,3 кПа? Если давление держится на уровне 50 кПа?
Показать решение В качестве другого примера рассмотрим фазовую диаграмму диоксида углерода, показанную на рисунке 5. Кривая твердое тело-жидкость имеет положительный наклон, что указывает на то, что температура плавления CO 2 увеличивается с давлением, как и для большинства веществ (вода является заметным исключением, как описано ранее). Обратите внимание, что тройная точка намного выше 1 атм, что указывает на то, что углекислый газ не может существовать в виде жидкости в условиях атмосферного давления. Вместо этого охлаждение газообразного диоксида углерода до 1 атм приводит к его осаждению в твердом состоянии. Точно так же твердый диоксид углерода не плавится при давлении 1 атм, а возгоняется с образованием газообразного CO 9 .0025 2 . Наконец, обратите внимание, что критическая точка для углекислого газа наблюдается при относительно скромных температуре и давлении по сравнению с водой.
Кривая твердое тело-жидкость имеет положительный наклон, что указывает на то, что температура плавления CO 2 увеличивается с давлением, как и для большинства веществ (вода является заметным исключением, как описано ранее). Обратите внимание, что тройная точка намного выше 1 атм, что указывает на то, что углекислый газ не может существовать в виде жидкости в условиях атмосферного давления. Вместо этого охлаждение газообразного диоксида углерода до 1 атм приводит к его осаждению в твердом состоянии. Точно так же твердый диоксид углерода не плавится при давлении 1 атм, а возгоняется с образованием газообразного CO 9 .0025 2 . Наконец, обратите внимание, что критическая точка для углекислого газа наблюдается при относительно скромных температуре и давлении по сравнению с водой.
Рис. 5. Показана фазовая диаграмма диоксида углерода. Ось давления построена в логарифмическом масштабе, чтобы соответствовать большому диапазону значений.
Пример 2:
Определение состояния углекислого газаИспользуя фазовую диаграмму для углекислого газа, показанную на рисунке 5, определите состояние CO 2 при следующих температурах и давлениях:
- −30 °C и 2000 кПа
- −60 °C и 1000 кПа
- −60 °C и 100 кПа
- 20 °C и 1500 кПа
- 0 °C и 100 кПа
- 20 °C и 100 кПа
Определите фазовые превращения, которые претерпит диоксид углерода при повышении его температуры с -100 °C при постоянном давлении 1500 кПа. При 50 кПа. При каких примерных температурах происходят эти фазовые превращения?
При 50 кПа. При каких примерных температурах происходят эти фазовые превращения?
Сверхкритические жидкости
Если мы поместим образец воды в герметичный контейнер при температуре 25 °C, удалив воздух и позволив установиться равновесию испарения-конденсации, мы получим смесь жидкой воды и водяного пара при давление 0,03 атм. Отчетливо наблюдается четкая граница между более плотной жидкостью и менее плотным газом. По мере повышения температуры давление водяного пара увеличивается, как описано кривой жидкость-газ на фазовой диаграмме для воды (рис. 2), и сохраняется двухфазное равновесие жидкой и газообразной фаз. При температуре 374 °С давление пара возросло до 218 атм, и дальнейшее повышение температуры приводит к исчезновению границы между жидкой и паровой фазами. Вся вода в сосуде теперь находится в одной фазе, физические свойства которой занимают промежуточное положение между газообразным и жидким состояниями. Эта фаза вещества называется сверхкритическая жидкость , а температура и давление, выше которых существует эта фаза, являются критической точкой . При температуре выше критической газ не может сжижаться, какое бы давление ни применялось. Давление, необходимое для сжижения газа при его критической температуре, называется критическим давлением. Критические температуры и критические давления некоторых распространенных веществ приведены в таблице 1.
При температуре выше критической газ не может сжижаться, какое бы давление ни применялось. Давление, необходимое для сжижения газа при его критической температуре, называется критическим давлением. Критические температуры и критические давления некоторых распространенных веществ приведены в таблице 1.
| Таблица 1. | ||
|---|---|---|
| Вещество | Критическая температура (К) | Критическое давление (атм) |
| водород | 33,2 | 12,8 |
| азот | 126,0 | 33,5 |
| кислород | 154,3 | 49,7 |
| двуокись углерода | 304,2 | 73,0 |
| аммиак | 405,5 | 111,5 |
| диоксид серы 9{\circ}{\text{C}}[/латекс]) | Критическое давление (кПа) | |
| водород | -240,0 | 1300 |
| азот | -147,2 | 3400 |
| кислород | -118,9 | 5000 |
| двуокись углерода | 31,1 | 7400 |
| аммиак | 132,4 | 11 300 |
| диоксид серы | 157,2 | 7800 |
| вода | 374,0 | 22 000 |
Рис. 6. (а) Герметичный контейнер с жидким диоксидом углерода нагревается немного ниже его критической точки, что приводит к (б) образованию сверхкритической флюидной фазы. Охлаждение сверхкритической жидкости снижает ее температуру и давление ниже критической точки, что приводит к восстановлению отдельных жидких и газообразных фаз (c и d). Цветные поплавки иллюстрируют разницу в плотности между жидким, газообразным и сверхкритическим флюидным состоянием. (кредит: модификация работы «mrmrobin»/YouTube)
6. (а) Герметичный контейнер с жидким диоксидом углерода нагревается немного ниже его критической точки, что приводит к (б) образованию сверхкритической флюидной фазы. Охлаждение сверхкритической жидкости снижает ее температуру и давление ниже критической точки, что приводит к восстановлению отдельных жидких и газообразных фаз (c и d). Цветные поплавки иллюстрируют разницу в плотности между жидким, газообразным и сверхкритическим флюидным состоянием. (кредит: модификация работы «mrmrobin»/YouTube)
В этом видео наблюдайте за переходом диоксида углерода из жидкости в сверхкритическую жидкость.
Вы можете просмотреть стенограмму «Сверхкритический CO2» здесь (откроется в новом окне).
Подобно газу, сверхкритическая жидкость будет расширяться и заполнять контейнер, но ее плотность намного выше плотности обычных газов и обычно близка к плотности жидкостей. Подобно жидкостям, эти жидкости способны растворять нелетучие растворенные вещества. Однако они практически не проявляют поверхностного натяжения и имеют очень низкую вязкость, поэтому они могут более эффективно проникать в очень маленькие отверстия в твердой смеси и удалять растворимые компоненты. Эти свойства делают сверхкритические жидкости чрезвычайно полезными растворителями для широкого спектра применений. Например, сверхкритический диоксид углерода стал очень популярным растворителем в пищевой промышленности, его используют для декофеинизации кофе, удаления жиров из картофельных чипсов и извлечения вкусовых и ароматических соединений из цитрусовых масел. Он нетоксичен, относительно недорог и не считается загрязнителем. После использования CO 2 можно легко восстановить, снизив давление и собрав образовавшийся газ.
Подобно жидкостям, эти жидкости способны растворять нелетучие растворенные вещества. Однако они практически не проявляют поверхностного натяжения и имеют очень низкую вязкость, поэтому они могут более эффективно проникать в очень маленькие отверстия в твердой смеси и удалять растворимые компоненты. Эти свойства делают сверхкритические жидкости чрезвычайно полезными растворителями для широкого спектра применений. Например, сверхкритический диоксид углерода стал очень популярным растворителем в пищевой промышленности, его используют для декофеинизации кофе, удаления жиров из картофельных чипсов и извлечения вкусовых и ароматических соединений из цитрусовых масел. Он нетоксичен, относительно недорог и не считается загрязнителем. После использования CO 2 можно легко восстановить, снизив давление и собрав образовавшийся газ.
Пример 3:
Критическая температура углекислого газа Если встряхнуть углекислотный огнетушитель в прохладный день (18 °C), можно услышать, как жидкий CO 2 плещется внутри цилиндра.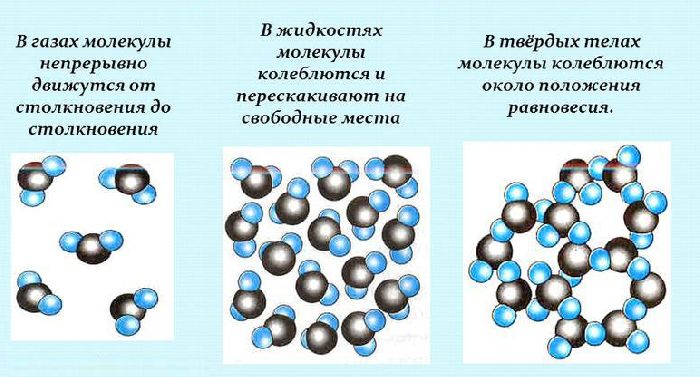 Однако тот же самый цилиндр не содержит жидкости в жаркий летний день (35 °C). Объясните эти наблюдения.
Однако тот же самый цилиндр не содержит жидкости в жаркий летний день (35 °C). Объясните эти наблюдения.
Аммиак можно сжижать путем сжатия при комнатной температуре; кислород не может быть сжижен в этих условиях. Почему два газа ведут себя по-разному?
Показать решениеДекофеинизация кофе с использованием сверхкритического CO
2Кофе является вторым наиболее широко продаваемым товаром в мире после нефти. Во всем мире люди любят аромат и вкус кофе. Многие из нас также зависят от одного компонента кофе — кофеина, который помогает нам двигаться утром или сохранять бодрость днем. Но в конце дня стимулирующий эффект кофе может помешать вам уснуть, поэтому вечером вы можете пить кофе без кофеина.
С начала 1900-х годов для удаления кофеина из кофе использовалось множество методов. У всех есть преимущества и недостатки, и все зависит от физических и химических свойств кофеина. Поскольку кофеин представляет собой несколько полярную молекулу, он хорошо растворяется в воде, полярной жидкости. Однако, поскольку многие из более чем 400 других соединений, влияющих на вкус и аромат кофе, также растворяются в H 2 O, процессы декофеинизации горячей водой также могут удалять некоторые из этих соединений, что отрицательно влияет на запах и вкус кофе без кофеина. Дихлорметан (СН 2 Cl 2 ) и этилацетат (CH 3 CO 2 C 2 H 5 ) имеют полярность, аналогичную кофеину, и поэтому являются очень эффективными растворителями для экстракции кофеина. но оба также удаляют некоторые компоненты вкуса и аромата, и их использование требует длительного времени экстракции и очистки. Поскольку оба этих растворителя являются токсичными, возникают опасения по поводу воздействия остаточного растворителя, остающегося в кофе без кофеина, на здоровье.
Поскольку кофеин представляет собой несколько полярную молекулу, он хорошо растворяется в воде, полярной жидкости. Однако, поскольку многие из более чем 400 других соединений, влияющих на вкус и аромат кофе, также растворяются в H 2 O, процессы декофеинизации горячей водой также могут удалять некоторые из этих соединений, что отрицательно влияет на запах и вкус кофе без кофеина. Дихлорметан (СН 2 Cl 2 ) и этилацетат (CH 3 CO 2 C 2 H 5 ) имеют полярность, аналогичную кофеину, и поэтому являются очень эффективными растворителями для экстракции кофеина. но оба также удаляют некоторые компоненты вкуса и аромата, и их использование требует длительного времени экстракции и очистки. Поскольку оба этих растворителя являются токсичными, возникают опасения по поводу воздействия остаточного растворителя, остающегося в кофе без кофеина, на здоровье.
Сверхкритическая флюидная экстракция с использованием диоксида углерода в настоящее время широко используется как более эффективный и экологически безопасный метод декофеинизации (рис. 7). При температуре выше 304,2 К и давлении выше 7376 кПа СО 2 — сверхкритическая жидкость, обладающая свойствами как газа, так и жидкости. Подобно газу, он проникает глубоко в кофейные зерна; подобно жидкости, он эффективно растворяет определенные вещества. Экстракция пропаренных кофейных зерен сверхкритическим диоксидом углерода удаляет 97-99% кофеина, оставляя вкусовые и ароматические соединения кофе нетронутыми. Поскольку CO 2 представляет собой газ при стандартных условиях, его удаление из экстрагированных кофейных зерен осуществляется легко, как и извлечение кофеина из экстракта. Кофеин, извлеченный из кофейных зерен с помощью этого процесса, является ценным продуктом, который впоследствии можно использовать в качестве добавки к другим продуктам питания или лекарствам.
7). При температуре выше 304,2 К и давлении выше 7376 кПа СО 2 — сверхкритическая жидкость, обладающая свойствами как газа, так и жидкости. Подобно газу, он проникает глубоко в кофейные зерна; подобно жидкости, он эффективно растворяет определенные вещества. Экстракция пропаренных кофейных зерен сверхкритическим диоксидом углерода удаляет 97-99% кофеина, оставляя вкусовые и ароматические соединения кофе нетронутыми. Поскольку CO 2 представляет собой газ при стандартных условиях, его удаление из экстрагированных кофейных зерен осуществляется легко, как и извлечение кофеина из экстракта. Кофеин, извлеченный из кофейных зерен с помощью этого процесса, является ценным продуктом, который впоследствии можно использовать в качестве добавки к другим продуктам питания или лекарствам.
Рис. 7. (a) Молекулы кофеина имеют как полярные, так и неполярные области, что делает его растворимым в растворителях различной полярности. (b) На схеме показан типичный процесс удаления кофеина с использованием сверхкритического диоксида углерода.
Ключевые понятия и резюме
Условия температуры и давления, при которых вещество существует в твердом, жидком и газообразном состояниях, представлены на фазовой диаграмме для этого вещества. Фазовые диаграммы представляют собой объединенные графики трех кривых равновесия давления и температуры: твердое тело-жидкость, жидкость-газ и твердое тело-газ. Эти кривые представляют отношения между температурами фазового перехода и давлениями. Точка пересечения всех трех кривых представляет тройную точку вещества — температуру и давление, при которых все три фазы находятся в равновесии. При давлениях ниже тройной точки вещество не может существовать в жидком состоянии независимо от его температуры. Конец кривой жидкость-газ представляет критическую точку вещества, давление и температуру, выше которых не может существовать жидкая фаза.
Попробуйте
- По фазовой диаграмме воды (рис. 2) определите состояние воды при:
- 35 °C и 85 кПа
- −15 °C и 40 кПа
- −15 °C и 0,1 кПа
- 75 °C и 3 кПа
- 40 °C и 0,1 кПа
- 60 °C и 50 кПа
- Какие фазовые превращения произойдут, если воду подвергнуть воздействию переменного давления при постоянной температуре 0,005 °С? При 40°С? При −40 °С?
- Скороварки позволяют готовить пищу быстрее, потому что более высокое давление внутри скороварки увеличивает температуру кипения воды.
 В некоторых скороварках есть предохранительный клапан, который выпускает пар, если давление превышает 3,4 атм. Какая приблизительная максимальная температура может быть достигнута внутри этой скороварки? Объясните свои рассуждения.
В некоторых скороварках есть предохранительный клапан, который выпускает пар, если давление превышает 3,4 атм. Какая приблизительная максимальная температура может быть достигнута внутри этой скороварки? Объясните свои рассуждения. - Из диаграммы состояния диоксида углерода на рисунке 5 определите состояние CO 2 при:
- 20 °C и 1000 кПа
- 10 °C и 2000 кПа
- 10 °C и 100 кПа
- −40 °C и 500 кПа
- −80 °C и 1500 кПа
- −80 °C и 10 кПа
- Определите фазовые превращения, которые претерпевает диоксид углерода при изменении давления, если поддерживать температуру на уровне -50 °C? Если поддерживать температуру минус 40 °С? При 20°С? (См. фазовую диаграмму на рисунке 5).
- Рассмотрим цилиндр, содержащий смесь жидкой двуокиси углерода, находящейся в равновесии с газообразной двуокисью углерода, при начальном давлении 65 атм и температуре 20 °С. Нарисуйте график, изображающий изменение давления в баллоне во времени при выделении газообразного диоксида углерода при постоянной температуре.
- Сухой лед, CO 2 ( s ), не тает при атмосферном давлении. Возгоняется при температуре -78°С. При каком минимальном давлении CO 2 ( s ) расплавится с образованием CO 2 ( l )? Примерно при какой температуре это произойдет? (См. фазовую диаграмму на рис. 5.)
- Если сильный шторм привел к отключению электричества, может потребоваться использование бельевой веревки для сушки белья. Во многих частях страны в разгар зимы одежда быстро замерзает, если ее повесить на веревку. Если не будет снега, они все равно высохнут? Поясните свой ответ.
- Можно ли сжижать азот при комнатной температуре (около 25 °C)? Можно ли сжижать диоксид серы при комнатной температуре? Объясните свои ответы.
- Элементарный углерод имеет одну газовую фазу, одну жидкую фазу и три различные твердые фазы, как показано на фазовой диаграмме:
- На фазовой диаграмме обозначьте газовую и жидкую области.
- Графит является наиболее стабильной фазой углерода при нормальных условиях. На фазовой диаграмме обозначьте графитовую фазу.
- Если графит при нормальных условиях нагреть до 2500 К при повышении давления до 10 5 атм, то он превращается в алмаз. Обозначьте алмазную фазу.
- Обведите каждую тройную точку на фазовой диаграмме.
- В какой фазе существует углерод при 4000 К и 10 5 атм?
- Если температуру образца углерода увеличить с 4000 К до 5000 К при постоянном давлении 10 2 атм, какой фазовый переход произойдет, если произойдет?
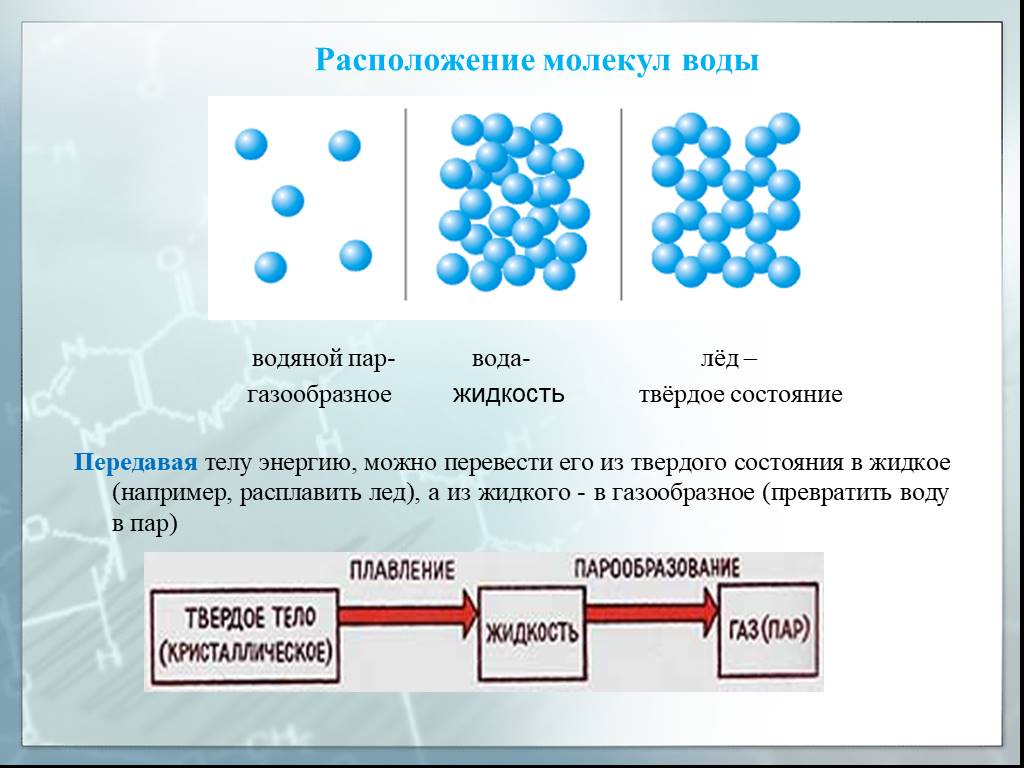 В некоторых скороварках есть предохранительный клапан, который выпускает пар, если давление превышает 3,4 атм. Какая приблизительная максимальная температура может быть достигнута внутри этой скороварки? Объясните свои рассуждения.
В некоторых скороварках есть предохранительный клапан, который выпускает пар, если давление превышает 3,4 атм. Какая приблизительная максимальная температура может быть достигнута внутри этой скороварки? Объясните свои рассуждения.
 На фазовой диаграмме обозначьте графитовую фазу.
На фазовой диаграмме обозначьте графитовую фазу.Глоссарий
критическая точка: температура и давление, выше которых газ не может сконденсироваться в жидкость
фазовая диаграмма: график давление-температура, обобщающий условия, при которых могут существовать фазы вещества
сверхкритическая жидкость: вещество при температуре и давлении выше его критической точки; проявляет свойства, промежуточные между газообразным и жидким состояниями
тройная точка: температура и давление, при которых паровая, жидкая и твердая фазы вещества находятся в равновесии
Состояние вещества
Состояние вещества Газы, жидкости и твердые тела состоят из микроскопических частиц, но поведение этих частиц различается в трех фазах. На следующем рисунке показаны микроскопические различия.
На следующем рисунке показаны микроскопические различия.
| Газ под микроскопом. | Жидкость под микроскопом. | Вид твердого тела под микроскопом. |
Обратите внимание, что:
- Частиц в:
- газ хорошо разделены без регулярного расположения.
- жидкости расположены близко друг к другу без регулярного расположения.
- твердые тела плотно упакованы, обычно в правильном порядке.
- Частиц в:
- газ вибрирует и свободно перемещается на высоких скоростях.
- жидкости вибрируют, перемещаются и скользят друг мимо друга.
- твердые вибрируют (покачиваются), но обычно не перемещаются с места на место.
