
Теория химического строения — Википедия
Теория химического строения — учение о строении молекулы, описывающее все те её характеристики, которые в своей совокупности определяют химическое поведение (реакционную способность) данной молекулы. Сюда относятся: природа атомов, образующих молекулу, их валентное состояние, порядок и характер химической связи между ними, пространственное их расположение, характерное распределение электронной плотности, характер электронной поляризуемости электронного облака молекулы и т. д.[1]
Основные положения теории химического строения, являющейся фундаментом химии, были развиты русским химиком А. М. Бутлеровым.
Бутлеров определял понятие химического строения так:
Исходя от мысли, что каждый химический атом, входящий в состав тела, принимает участие в образовании этого последнего и действует здесь определённым количеством принадлежащей ему химической силы (сродства), я называю химическим строением распределение действия этой силы, вследствие которой химические атомы, посредственно или непосредственно влияя друг на друга, соединяются в химическую частицу
[1].
Предпосылки создания теории химического строения[править | править код]
В 1812 году итальянский физик и химик Амедео Авогадро, изучая молекулярные веса газов (водорода, кислорода, азота, хлора), выдвинул молекулярную гипотезу строения вещества. Однако работа Авогадро долгое время не получала признания, что тормозило развитие основных идей в области химического строения молекул. Лишь после убедительного доклада Станислао Канниццаро на первом международном съезде химиков в Карлсруэ (1860) атомные веса, определённые с помощью закона Авогадро, стали общепринятыми[2]. На съезде разграничили понятия «атом», «молекула», утвердили атомно-молекулярное учение, основное положение которого «атомы при взаимодействии образуют молекулу».
Атомно-молекулярное учение послужило основой создания теории химического строения Бутлерова.
Теория химического строения Бутлерова[править | править код]
Термин «химическое строение» впервые ввёл А. М. Бутлеров 19 сентября 1861 года в докладе «О химическом строении веществ» на химической секции Съезда немецких естествоиспытателей и врачей в Шпейере (опубликованном в том же году на немецком[3] и в следующем — на русском языках[4]). В том же докладе он заложил основы классической теории химического строения. Главные положения этой теории следующие:
- атомы в молекулах веществ соединены друг с другом согласно их валентности, порядок распределения связей в молекуле называется химическим строением;
- изменение этой последовательности приводит к образованию нового вещества с новыми свойствами;
- свойства веществ зависят не только от их качественного и количественного состава, но и от «химического строения», то есть от порядка соединения атомов в молекулах и характера их взаимного влияния. Наиболее сильно влияют друг на друга атомы, непосредственно связанные между собой;
- атомы в молекулах оказывают влияние друг на друга, и это влияние приводит к химическим изменениям поведения атома;
- определить состав и строение химического вещества можно по продуктам химических превращений.
В 1864 году Бутлеров первым объясняет явление изомерии, показав что изомеры — это соединения, обладающие одинаковым элементным составом, но различным химическим строением. В 1874 году возникает стереохимия, или трёхмерная структурная химия в форме постулата Вант-Гоффа о тетраэдрической системе валентностей у атома углерода.
В настоящее время принято различать структурную и пространственную изомерию.
Структурную изомерию подразделяют на изомерию скелета, обусловленную различным порядком связи атомов, образующих скелет молекулы, например в Н-бутане и изобутане, и на изомерию положения одинаковых функциональных групп при одинаковом углеродном скелете молекулы, например в орто- , мета- и пара- изомерах ароматических соединений.
Пространственная изомерия обусловлена существованием стереоизомеров, соединений, имеющих одинаковый порядок связей атомов, но различное пространственное расположение. К видам пространственной изомерии относятся: оптическая изомерия, обусловливающая существование энантиомеров — пары стереоизомеров, представляющих собой зеркальные отражения друг от друга, не совмещаемые в пространстве; диастереомерия, обусловливающая существование изомеров, не являющихся энантиомерами; геометрическая изомерия, обусловливающая цис- и транс- изомеров, свойственных соединениям с двойными связями и малыми циклами.
Электронные интерпретации строения молекул[править | править код]
Строение молекулы H2 по БоруВ 1913 году датский физик Нильс Бор предложил рассматривать электронную пару в форме вращающегося кольца, плоскость которого перпендикулярна оси молекулы и равноудалена от ядер атомов.
Динамическое равновесие молекулярной системы, по Бору, достигается за счёт баланса сил между силами взаимного отталкивания ядер и силами притяжения ядер к плоскости кольца электронов. Боровская интерпретация строения молекулы учитывала кулоновское отталкивание электронов, в кольце они находятся в диаметрально противоположном положении.
Молекулу метана Нильс Бор описывал следующим образом:
Схема диполь-дипольного взаимодействия двух атомов водорода по ЛондонуЯдро углерода, заключённое в очень маленькое кольцо из двух электронов, расположено в центре (тетраэдра), а ядро водорода по углам. Химические связи представляют собой четыре двухэлектронных кольца, вращающихся вокруг линий, соединяющих центр с углами
[5].
В своём докладе, прочитанном в Королевском колледже в Лондоне в ознаменовании столетия опубликования трудов Дж. Максвелла по электромагнитному излучению, К. Коулсон дал анализ происхождения и сущности межатомных сил, приводящих к образованию молекулы[6]. Коулсон, ссылаясь на работу Лондона, на примере двух атомов водорода показывает «каким образом два нейтральных атома или молекулы могут оказывать притяжение друг к другу на значительном расстоянии».
Ядра A и B двух атомов водорода находятся на расстоянии r друг от друга (рис.). Каждый атом несёт по одном электрону (в P и Q соответственно). Совокупность зарядов +e в A и -e в P приблизительно эквивалентна электрическому диполю, имеющему величину e·AP. Подобным же образом совокупность +e в B и -e в Q приблизительно эквивалентна электрическому диполю e·BQ. Эти два диполя взаимодействуют друг с другом. Общая потенциальная энергия двух диполей, m и m’, находящихся на расстоянии r, равна:
mm′r3−3(mr)(m′r)r5{\displaystyle {\frac {mm’}{r^{3}}}-3{\frac {(mr)(m’r)}{r^{5}}}}[6]
На языке волновой механики это выражение рассматривается как возмущение, действующее на оба атома.
Природа межатомных сил имеет электромагнитный характер и называется диполь-дипольным взаимодействием. Существуют диполь-квадрупольные, квадруполь-квадрупольные и другие взаимодействия, при которых энергия изменяется в зависимости от более высоких степеней 1/r[6].
Распределение электронной плотности в химических соединениях[править | править код]
 Рис. Радиальное распределение вероятности нахождения электрона в атоме водорода.
Рис. Модель молекулы водорода и её осевая проекция
Рис. Радиальное распределение вероятности нахождения электрона в атоме водорода.
Рис. Модель молекулы водорода и её осевая проекцияЭлектронная плотность — это плотность вероятности обнаружения электрона в данной точке пространства. Электронная плотность нормирована и, соответственно, указывает на вероятное число электронов в данном элементарном объёме.
Вероятностную (статистическую) интерпретацию волновой функции сформулировал М. Борн в 1926 году как только было опубликована волновое уравнение Шрёдингера. В 1954 году М. Борн удостоен Нобелевской премии по физике с формулировкой «За фундаментальное исследование в области квантовой механики, особенно за статистическую интерпретацию волновой функции».
Расчёт электронной плотности проводят с использованием уравнения Шрёдингера, которое решается аналитически точно для систем, содержащих только один электрон[7].
Получаемая радиальная функция распределения вероятности нахождения электрона в атоме водорода обладает максимумом при α0, как показано на рисунке. Этот наиболее вероятный радиус совпадает с боровским радиусом и принят в качестве атомной единицы для линейных размеров 1 а. е. (бор) = 0,529177·10−10м ≈ 0,529 Å. Более размытое облако плотности вероятности, полученное при решении уравнения Шрёдингера для атома водорода, значительно отличается от боровской модели атома и согласуется с принципами неопределённости Гейзенберга. С учётом статистической интерпретации волновой функции М. Борна и принципа неопределённости Гейзенберга длины диполей AP и BQ взаимодействующих атомов в расчётах Ф. Лондона достаточно размыты. Размыто и электронное кольцо в модели молекулы водорода по Н. Бору до состояния тороидального электронного облака с неопределёнными границами.
Наиболее вероятный радиус электронного кольца (тора) молекулы водорода (re) определяется боровским радиусом (α0) и длиной химической связи (d): re2 = α02 — (d/2)2; re = 0,374 Å.
Благодаря пространственной симметрии дипольный момент молекулы водорода равен нулю, что соответствует её низкой химической активности[8]. Пространственная симметрия электронной плотности сохраняется, если соединяющиеся в молекулу атомы имеют одинаковую энергию ионизации. В этом случае связывающее электронное кольцо располагается на равном расстоянии от каждого из ядер. Если же потенциалы ионизации различны электронная плотность смещается в сторону атома с большим первым потенциалом ионизации[9]. Смещение электронной плотности приводит к асимметрии распределения электрических зарядов в молекуле, молекула становится полярной системой с определённым дипольным моментом.
Приближённые квантовохимические методы расчёта[править | править код]
Поскольку точное решение уравнения Шрёдингера для атомно-молекулярных систем, содержащих два и более электрона, невозможно, предложены приближённые методы расчёта электронной плотности. Все они возникли в 1930-х годах, проделали значительный путь развития и долгое время дополняли друг друга. Наиболее важные из них — теория валентных связей, теория молекулярных орбиталей, теория кристаллического поля, теория функционала плотности.
В рамках теории валентных связей разработана концепция резонанса (Л. Полинг) и родственная ей концепция мезомерии (К. Ингольд). Концепция резонанса рассмотрена на примере молекулярного иона водорода. Л. Полинг показал, что стабильность молекулярного иона водорода:
вызвана резонансом электрона, то есть движением его «взад и вперёд» между двумя ядрами с «резонансной частотой», равной по величине энергии резонанса (50 ккал/моль), делённой на константу Планка h. Для молекулярного иона в основном состоянии эта частота равна 7.1014сек−1[10].
Концепция резонанса дополняла постулаты классической теории химического строения и утверждала, что если для данного соединения классическая теория допускает построение нескольких приемлемых структурных формул, то действительному состоянию молекул этого соединения отвечает не какая-либо одна отдельная формула, а некоторое их сочетание (наложение, резонанс структур)[11].
Мезомерия является теорией электронного строения химических соединений, согласно которой истинное распределение электронной плотности в молекуле является промежуточным между распределениями, представленными несколькими классическими формулами[12].
Обычно рассматривают положительный и отрицательный мезомерные эффекты:
Винилхлорид: +М-эффект
Аллил-катион: −М-эффект
Кампания идеологического вмешательства в теорию химического строения[править | править код]
Кампания началась в 1949 году с публикации статьи В. М. Татевского и М. М. Шахпаронова «Об одной махистской теории в химии и её пропагандистах»[13]. В качестве главного объекта нападения была выбрана теория резонанса Л. Полинга. Было объявлено, что «представления о реальной молекуле как о чём-то среднем между двумя (и более) крайними абстрактными структурами являются буржуазными и поэтому направленными против всего самого „святого“». Были указаны и пропагандисты теории Я. К. Сыркин и М. Е. Дяткина — авторы книги «Химическая связь и строение молекул», в которой нашла отражение теория резонанса.
В воздухе очередной раз запахло инквизицией. В этой тревожной обстановке ведущие химики страны собрались на Всесоюзное совещание по проблемам химического строения (1951 г., Москва). Стенограмма этого совещания — один из наиболее позорных документов, когда-либо созданных коллективными усилиями ученых, хранится в химических библиотеках всего мира, и от этого срама бог весть когда ещё удастся отмыться… До крови не дошло — спасла оттепель, начавшаяся весной следующего года. Я. К. Сыркин и М. Е. Дяткина, подготовленные недавними друзьями и коллегами к выдаче в качестве первых козлов отпущения, уцелели; более того Я. К. Сыркин в дальнейшем стал академиком[14].
Лайнус Полинг удостоен в 1954 году Нобелевской премии по химии «за исследование природы химической связи и её применение для определения структуры сложных соединений».
Однако консенсус в теории химического строения не был достигнут. В. М. Татевский в курсе «Строение молекул» (1977 г.) отмечал:
… полностью выпадают и «висят в воздухе» представления уходящих в прошлое, но всё ещё фигурирующих в литературе так называемых «теории резонанса» и «теории мезомерии», которые не имеют основы ни в классической теории химического строения, ни в законных приложениях классической физики к вопросам строения молекул, ни в квантовой механике[15].
Лишь в 1991 году проведён принципиальный анализ кампании по борьбе с теорией резонанса и было показано, что эта кампания «нанесла серьёзный ущерб престижу советской науки»[16].
В классической теории химического строения понятие атома в молекуле является изначальным. Интуитивно ясно, что атом в молекуле меняется, меняются и его свойства в зависимости от окружения этого атома, прежде всего ближайшего. Основным показателем является расстояние между атомами в молекуле, определяющее как длину химической связи, так и прочность химической связи.
В квантовой теории понятие атома вторично. Так, по утверждению В. М. Татевского, молекула не состоит из атомов: «С современной точки зрения ясно, что при образовании молекулы сохраняются не атомы, а только ядра атомов и электроны»[17].
Было предпринято множество попыток сохранить понятие атома в молекуле, но, тем не менее, практически всегда они не удовлетворяли последующих исследователей по тем или иным причинам.
Одна из наиболее удачных попыток сохранения классической концепции атома в молекуле принадлежит Р. Бейдеру и его сотрудникам[18]. В рамках этой теории (QTAIM) электронная плотность «задаёт некоторое скалярное поле в трёхмерном пространстве, которое может быть охарактеризовано, например, его совокупностью экстремальных точек, линий и поверхностей, особых точек и т. п.»[19].
Таким образом, в квантовой теории атомов в молекулах Р. Бейдера оказывается возможным физическое обоснование ключевых понятий химии «атом», «молекула», «химическая связь» в терминах топологии функции электронной плотности в трёхмерном пространстве и описание химического строения молекул.
Электронная корреляция и конфигурация молекул[править | править код]
Электронная корреляция (взаимная обусловленность движения всех электронов атомной или молекулярной системы как целого определяется электростатическим отталкиванием электронов и статическим особенностями систем, в частности принципом Паули (фермиевская корреляция). Полный учёт электронной корреляции при расчёте энергии и определении электронной структуры системы достигается методом конфигурационного взаимодействия.
Простая и надёжная система правил для понимания и предсказания конфигурации молекул заложена в теории отталкивания электронных пар, наиболее важное правило которой достаточно эмпирично, хотя имеет квантовомеханическое обоснование, заключающееся в принципе Паули, а именно «электронные пары принимают такое расположение на валентной оболочке атома, при котором они максимально удалены друг от друга». При этом конфигурация молекулы будет определяться числом связывающих и неподелённых электронных пар у центрального атома:
- две связывающие электронные пары дают линейную конфигурацию молекулы;
- три — конфигурацию правильного треугольника;
- четыре — конфигурацию тетраэдра;
- пять — тригонально-бипирамидальную конфигурацию;
- шесть — конфигурацию октаэдра.
Наличие неподелённых электронных пар у центрального атома приводит к расширению типов конфигураций молекулы[20].
Строение соединений благородных газов[править | править код]
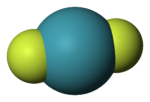 Молекула дифторида ксенона
Молекула дифторида ксенона 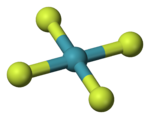
 Молекула гексафторида ксенона
Молекула гексафторида ксенонаОткрытие Н. Бартлеттом в 1962 году первого соединения ксенона положило начало интенсивному развитию химии благородных газов, что привело к получению большого ряда соединений инертных газов и установлению их химического строения.
Оказалось, что все соединения благородных газов имеют обыкновенные ковалентные связи и общепринятые конфигурации молекул. Так, конфигурация молекулы дифторида ксенона линейна: связи ксенон—фтор являются аксиальными и взаимодействуют с тремя неподелёнными электронными парами, находящимися в экваториальных положениях.
В молекуле тетрафторида ксенона осуществляется конфигурация с плоским квадратным расположением лигандов. Неподелённые электронные пары ксенона максимально удалены друг от друга и расположены по разные стороны от этого квадрата.
Структура молекулы гексафторида ксенона представляет собой искажённый октаэдр. «Отклонение атомов фтора от вершин правильного октаэдра указывает на то, что неподелённая электронная пара занимает положение над центром одной из граней октаэдра, а атомы фтора, расположенные в углах этой грани, вынуждены раздвинуться»[20].
Открытие Бартлетта показало ошибочность популярного в то время правила октета,
Строение электронодефицитных соединений[править | править код]
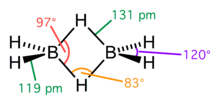
Структурная проблема, связанная с электронодефицитными соединениями, довольно сложна. Фундаментальная трудность заключалась в том, что в молекулах нет достаточного числа валентных электронов для того, чтобы связать все атомы обычными двухэлектронными связями. Так, например, в молекуле диборана имеется двенадцать валентных электронов, все двенадцать нужны для образования ковалентных связей шести атомов водорода с бором, так что для связи атомов бора между собой электронов не остаётся. Сам Полинг допускал, что в диборане функционируют одноэлектронные связи, а молекула в основном состоянии резонирует между семью структурами льюисовского типа, а также между многочисленными структурами, содержащими одноэлектронные связи.
Однако заслуженное признание получили исследования природы химической связи в бороводородах, выполненные американским физикохимиком У. Липскомбом. В его интерпретации в диборане имеют место четыре двух- и две трёхцентровые связи.
Четыре концевые двухцентровые двухэлектронные связи HB лежат в одной плоскости. Два же центральных атома водорода расположены симметрично над этой плоскостью и под нею и объединены с атомами бора двумя трёхцентровыми связями.
В 1976 году Липскомб удостоен Нобелевской премии по химии с формулировкой «за исследование структуры боранов (боргидритов), проясняющих проблему химических связей».
Особенности строения сэндвичевых соединений[править | править код]
Молекула ферроцена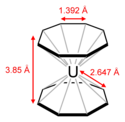
Дальнейшее развитие теории химического строения связано с открытием и установлением структуры ферроцена[21]. Оказалось, что при взаимодействии хлорида двухвалентного железа и циклопентадиена вместо ожидаемого соединения с двумя химическими связями железо-углерод образуется сэндвичеобразное соединение, в котором все 10 атомов углерода соединены с атомом железа.
Рентгеноструктурными исследованиями Э. Фишера установлено, что все атомы углерода в молекуле ферроцена структурно и химически эквивалентны. Атом металла взаимодействует не с одним или двумя конкретными атомами углерода, а со всеми атомами углерода двух циклопентадиенильных колец сразу. Атом металла как бы завис в пространстве между двумя циклами, представленных параллельными правильными пятиугольниками.
В настоящее время получены металлоцены — бициклопентадиенильные соединения переходных металлов для большинства d-элементов.
Сэндвичевые соединения образуются также с использованием в качестве органического фрагмента бензола или циклооктатетраена. Так, например, в ураноцене (см. рисунок) атом урана удерживает два восьмичленных кольца. Все 16 связей уран—углерод в ураноцене идентичны.
В 1973 году Э. Фишер и Дж. Уилкинсон удостоены Нобелевской премии по химии с формулировкой «За новаторскую, проделанную независимо друг от друга, работу в области металлорганических, так называемых сэндвичевых соединений».
Считается, что в сэндвичевых соединениях реализуется многоцентровая связь, тип химической связи, в которой в отличие от двухцентровой двухэлектронной связи, связывающие электронные пары распределены в пространстве трех или большего числа атомных центров молекулы, иона, радикала.[22]
- ↑ 1 2 Совещание по теории химического строения в органической химии. Успехи физических наук.. — 1951, октябрь. — Т. XLV, вып. 2. — С. 277—392.
- ↑ Химический энциклопедический словарь / гл. ред. И. Л. Кнунянц. — Сов. энциклопедия, 1983. — С. 652. — 792 с.
- ↑ Butlerow A. Einiges über die chemische Structur der Körper. (Vorgetragen in der chemischen Section der 36. Versammlung deutscher Naturforscher und Aerzte zu Speyer am 19.Septbr.) (нем.) // Zeitschrift für Chemie und Pharmacie : magazin. — 1861. — Bd. 4. — S. 549—560.
- ↑ Бутлеров А. О химическом строении веществ // Учёные записки Казанского университета (отд. физ.-мат. и мед. наук). Вып.1, отд.1. — 1862. — С. 1—11.
- ↑ Бор Н. Избранные научные труды. — М.: «Наука», 1970. — Т. 1. — С. 147. — 584 с.
- ↑ 1 2 3 Коулсон К. Успехи физических наук (статья «Межатомные силы — от Максвелла до Шрёдингера»). — 1963, ноябрь. — Т. LXXXI, вып.3. — С. 545-556.
- ↑ Шусторович Е. М. Химическая связь. Сущность и проблемы.. — М.: «Наука», 1973. — С. 6. — 232 с.
- ↑ Потапов А. А. Ренессанс классического атома. — М.: «Наука», 2011. — С. 352. — 444 с.
- ↑ Ганкин В. Ю., Ганкин Ю. В. Как образуется химическая связь и протекают химические реакции. — М.: «Граница». — С. 65. — 320 с.
- ↑ Паулинг Л. Природа химической связи. — М.-Л.: Химическая литература, 1947. — С. 25—26. — 440 с.
- ↑ Новая иллюстрированная энциклопедия. Пр-Ро. Кн.15. — М.: Большая российская энциклопедия, 2002. — С. 169. — 255 с.
- ↑ Новая иллюстрированная энциклопедия. Ма-Мо. Кн.11. — М.: Большая российская энциклопедия, 2002. — С. 141. — 255 с.
- ↑ Вопросы философии. — 1949. — № 3. — С. 176—192.
- ↑ Охлобыстин О. Ю. Жизнь и смерть химических идей. Очерки по истории теоретической химии.. — М.: Наука, 1989. — С. 184. — 190 с.
- ↑ Татевский В. М. Строение молекул.. — М.: Химия, 1977. — С. 10. — 512 с.
- ↑ Сонин А. С. Печальный юбилей одной кампании. (рус.) // Вестник РАН. — 1991. — Т. 61, № 8. — С. 96—107. Архивировано 5 мая 2010 года.
- ↑ Татевский В. М. Квантовая механика и теория строения молекул. — М.: Издательство МГУ, 1965. — С. 17. — 162 с.
- ↑ Бейдер Р. Атомы в молекулах. Квантовая теория. — М.: Мир, 2001. — 532 с.
- ↑ Степанов Н. Ф. Квантовая механика и квантовая химия. — М.: Мир, 2001. — С. 487. — 519 с.
- ↑ 1 2 Р. Гиллеспи. Геометрия молекул. — М.: Мир, 1975. — 278 с.
- ↑ Несмеянов А.Н. Ферроцен и родственные соединения. Избранные труды 1969 — 1979. — М.: Советская энциклопедия, 1982.
- ↑ Химический Энциклопедический словарь. — М.: Советская энциклопедия, 1983. — 792 с.
Cтроение молекул
Строение молекул
Молекула — наименьшая частица вещества, состоящая из одинаковых или различных атомов, соединенных между собой химическими связями, и являющаяся носителем его основных химических и физических свойств. Химические связи обусловлены взаимодействием внешних, валентных электронов атомов. Наиболее часто в молекулах встречается два типа связи: ионная и ковалентвая.
Ионная связь (например, в молекулах NаСl, КВr) осуществляется электростатическим взаимодействием атомов при переходе электрона одного атома к другому, т. е. при образовании положительного и отрицательного ионов. Ковалентная связь (например, в молекулах H2, С2, СО) осуществляется при обобществлении валентных электронов двумя соседними атомами (спины валентных электронов должны быть антипараллельны). Ковалентная связь объясняется на основе принципа неразличимости тождественных частиц, например электронов в молекуле водорода. Неразличимость частиц приводит к специфическому взаимодействию между ними, называемому обменным взаимодействием. Это чисто квантовый эффект, не имеющий классического объяснения, но его можно себе представить так, что электрон каждого из атомов молекулы водорода проводит некоторое время у ядра другого атома и, следовательно, осуществляется связь обоих атомов, образующих молекулу. При сближении двух водородных атомов до расстояний порядка боровского радиуса возникает их взаимное притяжение и образуется устойчивая молекула водорода.
Молекула является квантовой системой; она описывается уравнением Шредингера, учитывающим движение электронов в молекуле, колебания атомов молекулы, вращение молекулы. Решение этого уравнения — очень сложная задача, которая обычно разбивается на две: для электронов и ядер.
Энергия изолированной молекулы
EЕэл+Екол+Евращ, (1)
где Еэл — энергия движения электронов относительно ядер, Екол — энергия колебаний
ядер (в результате которых периодически изменяется относительное положение ядер), Евращ — энергия вращения ядер (в результате которых периодически изменяется ориентация молекулы в пространстве). В формуле (1) не учтены энергия поступательного движения центра масс молекулы и энергия ядер атомов в молекуле. Первая из них не квантуется, поэтому ее изменения не могут привести к возникновению молекулярного спектра, а вторую можно не учитывать, если не рассматривать сверхтонкую структуру
спектральных линий. Отношения Еэл:Екол:Евращ=1: m/M , где т — масса электрона, М — величина, имеющая порядок массы ядер атомов в молекуле, m/M10-5 10-3. Поэтому Еэл>>Екол>>Евращ. Доказано, что Еэл1 10 эВ, Екол10-2 10-1 эВ, Евращ10-5 10-3 эВ.
Каждая из входящих в выражение (1) энергий квантуется (ей соответствует набор дискретных уровней энергии) и определяется квантовыми числами. При переходе из одного энергетического состояния в другое поглощается или испускается энергия E=h. При таких переходах одновременно изменяются энергия движения электронов, энергии колебаний и вращения. Из теории и эксперимента следует, что расстояние между вращательными уровнями энергии Евращ гораздо меньше расстояния между колебательными уровнями Екол, которое, в свою очередь, меньше расстояния между электронными уровнями Еэл. На рис. 1 схематически представлены уровни энергии двухатомной молекулы (для примера рассмотрены только два электронных уровня — показаны жирными линиями).
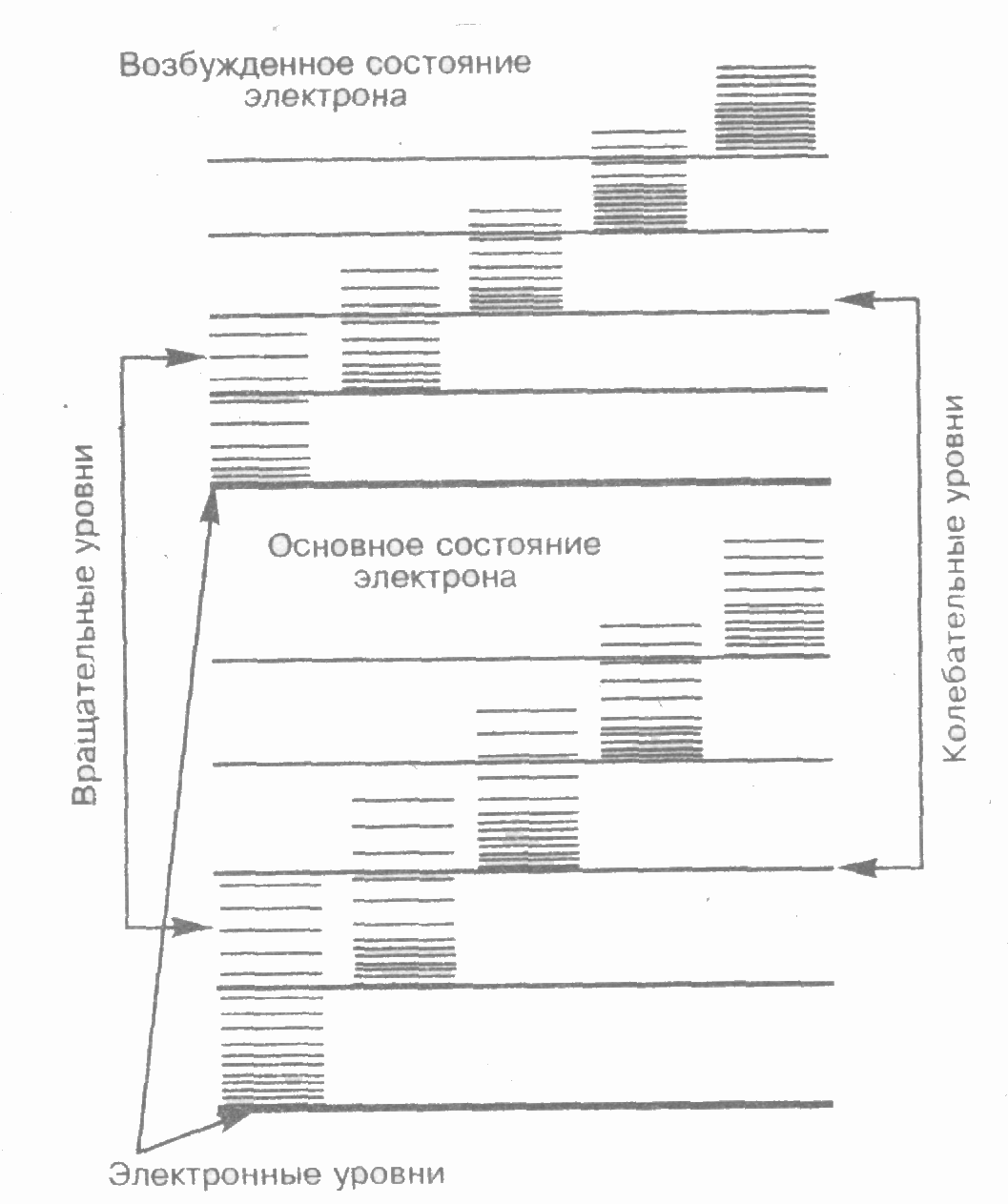 Рис.1
Рис.1
Как будет показано далее, структура энергетических уровней молекул определяет их спектр излучения, возникающий при квантовых переходах между соответствующими энергетическими уровнями.
Молекулярные спектры. Комбинационное рассеяние света
Строение молекул и свойства их энергетических уровней проявляются в молекулярных спектрах — спектрах излучения (поглощения), возникающих при квантовых переходах между уровнями энергии молекул. Спектр излучения молекулы определяется структурой ее энергетических уровней и соответствующими правилами отбора (так, например, изменение квантовых чисел, соответствующих как колебательному, так и вращательному движению, должно быть равно ± 1).
Итак, при разных типах переходов между уровнями возникают различные типы молекулярных спектров. Частоты спектральных линий, испускаемых молекулами, могут соответствовать переходам с одного электронного уровня на другой (электронные спектры) или с одного колебательного (вращательного) уровня на другой (колебательные (вращательные) спектры). Кроме того, возможны и переходы с одними значениями Екол и Евращ на уровни, имеющие другие значения всех трех компонентов, в результате чего возникают электронно-колебательные и колебательно-вращательные спектры. Поэтому спектр молекул довольно сложный.
Типичные молекулярные спектры — полосатые, представляющие собой совокупность более или менее узких полос в ультрафиолетовой, видимой и инфракрасной областях. Применяя спектральные приборы высокой разрешающей способности, можно видеть, что полосы представляют собой настолько тесно расположенные линии, что они с трудом разрешаются. Структура молекулярных спектров различна для разных молекул и с увеличением числа атомов в молекуле усложняется (наблюдаются лишь сплошные широкие полосы). Колебательными и вращательными спектрами обладают только многоатомные молекулы, а двухатомные их не имеют. Это объясняется тем, что двухатомные молекулы не имеют дипольиых моментов (при колебательных и вращательных переходах отсутствует изменение дипольиого момента, что является необходимым условием отличия от нуля вероятности перехода).
В 1928 г. академики Г. С. Ландсберг (1890—1957) и Л. И. Мандельштам и одновременно индийские физики Ч. Раман (1888—1970) и К. Кришнан (р. 1911) открыли явление комбинационного рассеяния света. Если на вещество (газ, жидкость, прозрачный кристалл) падает строго монохроматический свет, то в спектре рассеянного света помимо несмещенной спектральной линии обнаруживаются новые линии, частоты которых представляют собой суммы или разности частоты падающего света и частот i собственных колебаний (или вращений) молекул рассеивающей среды.
Линии в спектре комбинационного рассеяния с частотами —i, меньшими частоты
падающего света, называются стоксовыми (или красными) спутниками, линии с частотами +i, большими ,—антистоксовыми (или фиолетовыми) спутниками. Анализ спектров комбинационного рассеяния приводит к следующим выводам: 1) линии спутников располагаются симметрично по обе стороны от несмещенной линии; 2) частоты i не зависят от частоты падающего на вещество света, а определяются только рассеивающим веществом, т. е. характеризуют его состав и структуру; 3) число спутников определяется рассеивающим веществом; 4) интенсивность антистоксовых спутников меньше интенсивности стоксовых и с повышением температуры рассеивающего вещества увеличивается, в то время как интенсивность стоксовых спутников практически от температуры не зависит.
Объяснение закономерностей комбинационного рассеяния света дает квантовая теория. Согласно этой теории, рассеяние света есть процесс, в котором один фотон поглощается и один фотон испускается молекулой. Если энергии фотонов одинаковы, то в рассеянном свете наблюдается несмещенная линия. Однако возможны процессы рассеяния, при которых энергии поглощенного и испущенного фотонов различны. Различие энергии фотонов связано с переходом молекулы из нормального состояния в возбужденное (испущенный фотон будет иметь меньшую частоту — возникает сто-ксов спутник) либо из возбужденного состояния в нормальное (испущенный фотон будет иметь большую частоту — возникает антистоксов спутник).
Рассеяние света сопровождается переходами молекулы между различными колебательными или вращательными уровнями, в результате чего и возникает ряд симметрично расположенных спутников. Число спутников, таким образом, определяется энергетическим спектром молекул, т. е. зависит только от природы рассеивающего вещества. Так как число возбужденных молекул гораздо меньше, чем число невозбужденных, то интенсивность антистоксовых спутников меньше, чем стоксовых. С повышением температуры число возбужденных молекул растет, в результате чего возрастает и интенсивность антистоксовых спутников.
Молекулярные спектры (в том числе и спектры комбинационного рассеяния света) Применяются для исследования строения и свойств молекул, используются в молекулярном спектральном анализе, лазерной спектроскопии, квантовой электронике и т. д.
Молекулы структура, определение — Справочник химика 21
Одно из важнейших свойств химической связи — ее насыщаемость. Вследствие насыщаемости связи молекулы имеют определенный состав и существуют в виде дискретных частиц с определенной структурой. [c.66]Вопрос о механизме высыхания масел еще не разрешен, но с определенностью установлено, что полимеризации протекают через промежуточное образование перекисей, каталитически облегчающих образование трехмерных молекул, структура которых пока не установлена. [c.243]
При алкилировании к молекулам углеводородов присоединяются алкильные радикалы. В результате получают молекулы с определенной структурой, обеспечивающей требуемые свойства топлив. [c.9]
До открытия электрона невозможно было понять природу химической связи. Правда, понятие о валентности существовало уже в 1852 г. и в эти же годы существовали некоторые представления о геометрических формах молекул. Вант Гофф и Лебель установили тетраэдрическую структуру атома углерода, а Вернер создал стереохимию комплексных ионов. Очевидно, для того чтобы молекула имела определенную геометрическую форму, должны существовать какие-то связывающие силы между ее частями. В структурных формулах такую химическую связь между связанными атомами изображали черточкой. Она указывала на существование связи, но, разумеется, не давала никакого описания ее природы. Незадолго до открытия электрона Аррениус предположил существование свободных ионов. На основе этого предположения были сделаны многочисленные попытки найти объяснение силам, связывающим атомы. Хотя эти попытки были неудачными, они содействовали представлению об электрическом заряде как основе образования связи. После открытия электрона стало возможно дальнейшее развитие теории связи. В течение немногих лет, основываясь на положительно и отрицательно заряженных атомах, было предлолобъяснений образованию связи, но почти не было попыток связать заряды атома с его строением. В 1916 г. Льюис предложил свою теорию валентности. С тех пор было много сделано в области применения математики в теории валентности, но в основе представления о химической связи лежит по-прежнему теория Льюиса. Согласно Полингу , химическая связь возникает между двумя атомами в том случае, если связывающая атомы сила настолько велика, что приводит к образованию достаточно устойчивого агрегата, чтобы обеспечить его существование в виде самостоятельной частицы. Обычно различают пять типов химической связи ионная, ковалентная, металлическая, связь, обусловленная силами Ван-дер-Ваальса, и водородная, причем три первых очень прочны. Все эти связи одинаково важны, но металлическая связь здесь не будет рассмотрена о ней можно прочесть в других источниках . [c.134]
При рассмотрении электронной задачи предполагают, что геометрия молекулы фиксирована. В ряде случаев она известна из эксперимента. При отсутствии соответствующих данных в задачу входит и поиск оптимальной геометрии, что особенно важно в теории межмолекулярных взаимодействий, при рассмотрении структуры промежуточного комплекса в теории химических реакций и в других задачах. При рассмотрении адиабатического приближения (гл. 2, 1) уже упоминалось, что электронные и ядерные переменные не всегда удается разделить. Однако и в этих случаях на первом этапе исследования при расчете электронных характеристик исходят из некоторой заданной геометрии молекулы. Оператор энергии атома и оператор энергии молекулы характеризуются определенными свойствами симметрии, а именно инвариантностью относительно линейных преобразований электронных переменных. При переходе от теории атома к теории молекул изменяется пространственная симметрия, что следует принять во внимание при классификации электронных состояний. [c.187]
Эти же отношения в еще более тонкой форме наблюдаются при образовании мультиферментных комплексов. Такой комплекс может состоять, например, из семи различных ферментов, причем каждый из них представляет собой большую белковую молекулу. Собираясь вместе, эти молекулы испытывают определенные изменения формы, так что в итоге образуется высокоактивная структура. Подобные процессы относятся к явлениям самосборки , в которых точное взаимное структурное и энергетическое соответствие фрагментов приводит к созданию динамических структур, наделенных новыми свойствами, отличными от свойств фрагментов. Если проследить, что зто за новые свойства, то можно заметить, что они обеспечивают развитие новых кодовых связей, как правило, имеющих меньший термодинамический эквивалент. [c.341]
Каталитической активностью обладает не вся молекула фермента, а лишь определенный ее участок, называемый активным центром. Активный центр соединяется с молекулой реагирующего вещества, образуя непрочное промежуточное соединение, способное к дальнейшим превращениям. При этом активный центр вступает в соединение только с теми молекулами, структура которых подобна структуре активного центра. Этим, по-видимому, объясняется специфичность действия ферментов. [c.302]
Для вещественных структур определенного уровня пустотой являются структуры нижних уровней (см. примечание 25). Так, для кристаллической решетки пустотой являются отде тьные сложные молекулы, для молекул —отдельные атомы и т. д. [c.201]
Линейные или разветвленные молекулы в определенных условиях могут быть химически связаны между собой поперечными мостиками из атомов или групп атомов, образуя сшитые молекулы. Увеличение количества поперечных мостиков приводит к неограниченной по величине пространственной, трехмерной структуре (рис. 82). [c.189]
По молярной рефракции можно установить структуру молекулы. Для определения структуры молекулы подбирают такую структурную формулу, для которой вычисленная молярная рефракция по уравнению (И, 9) равна экспериментально полученному значению. [c.84]
Кроме цепных молекул, в определенных технологических условиях или в результате специальной обработки могут быть получены полимеры с пространственной структурой. Конечно, понятие молекулы у трехмерных полимеров теряет смысл, так как пространственные сетки могут достигать весьма больших размеров. Типичным примером полимера с пространственной структурой может служить вулканизованный каучук, состоящий из каучуковых молекул, сшитых друг с другом серными мостиками, [c.426]
Согласно исследованиям А. Приходько с сотрудниками твердый кислород при сверхнизких температурах имеет три кристаллических состояния, которые отличаются друг от друга взаимным расположением молекул. При определенной температуре структура кристалла резко меняется, имеются точки перехода . [c.174]
Почти идеальную возможность установ
14.2 Состав и строение молекул
Состав молекул приводится в виде химических формул. Химическая формула показывает, какие элементы и в каком количестве входят в состав данной молекулы (химического соединения). Следует различать простейшую и истинную формулу вещества. Простейшая формула показывает наименьшие целочисленные количества элементов в молекуле. Состав молекулы определяется валентностями элементов в данной молекуле. Например, формулы гидридов для элементов второго периода имеют следующий состав:
Li(I)Н(I), Ве(II)Н2, В(III)Н3, С(IV)Н4, N(III)Н3, Н2О(II), НF(I).
Строение молекул определяется строением атомов, входящих в состав данной молекулы, и геометрией орбиталей, образовавших химические связи. Строение молекул воды, аммиака и метана приведено далее на рисунке 16.1.
Если атом образует несколько связей с участием различных орбиталей, то может происходить такое явление, как гибридизация атомных орбиталей.
Гибридизация – это выравнивание формы и энергии различных орбиталей при образовании ковалентной связи.
Разберем данный вопрос на примере гибридизации орбиталей атома
углерода в молекуле метана (СН4). На рисунке 14.1 приведены четыре орбитали атома углерода и показаны формы четырех гибридных орбиталей. Так как в гибридизации участвуют одна s- и три p-орбитали, возникают четыре sp3-гибридные орбитали.
109о 28̓
s-, px-, py-, pz— орбитали атома углерода четыре sp3-гибридные орбитали
Рисунок 14.1 – sp3-гибридизация валентных орбиталей углерода
Четыре гибридные орбитали углерода располагаются симметрично относительно друг от друга. В результате этого молекула метана имеет форму правильного тетраэдра, у которого в центре находится атом C, а в вершинах – атомы H. Углы между всеми связями равны и составляют 109°28′.
15 Типы кристаллических решеток
Кристаллическая структура вещества характеризуется правильным (регулярным) расположением частиц в строго определенных местах в кристалле. Точки, в которых размещены частицы, называются узлами кристаллической решетки. В зависимости от того, какой тип взаимодействия осуществляется между частицами, занимающими узлы в кристаллической решетке, различают четыре типа кристаллических решеток: атомная, ионная, металлическая и молекулярная.
Атомная кристаллическая решётка – построена из атомов, соединенных между собой прочными ковалентными связями. Данные кристаллы обладают высокой температурой плавления и низкой тепло- и электропроводностью. Являются крайне твердыми, но хрупкими веществами. Например, алмаз.
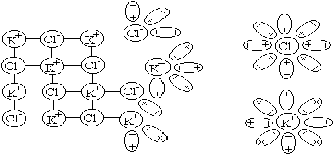
Ионная кристаллическая решётка состоит из положительных и отрицательных ионов, между которыми действуют электростатические силы. Температуры плавления ионных кристаллов выше, чем атомных и молекулярных. Такие кристаллы образуются между элементами с сильно различающимися электроотрицательностями. Например, NaCl.
Металлическая кристаллическая решетка – содержит в узлах кристаллической решётки ионы металла и свободные электроны, принадлежащие всему кристаллу металла. Взаимодействие между ионами металла и свободными электронами обеспечивает прочную металлическую связь. Свободные электроны могут свободно перемещаться в объёме кристалла, поэтому их иногда называют «электронным газом». Наличие электронов, свободно перемещающихся по всему кристаллу металла, объясняет такие характерные для металлов свойства, как высокие электро- и теплопроводность, пластичность.
Молекулярная кристаллическая решетка образуется между неполярными или слабополярными молекулами. Поскольку силы взаимодействия между молекулами в этих решетках являются слабыми, такие вещества плавятся при низких температурах. Большая часть веществ, которые при комнатной температуре находятся в жидком и газообразном состоянии, при низких температурах образуют молекулярные кристаллы. Например, СН4, СО2 и др. Энергия межмолекулярного взаимодействия меньше водородной связи и составляет примерно 2÷20 кДж/моль.
В 1873 г. голландский ученый Ван-дер-Ваальс объяснил природу сил, обуславливающих притяжение между отдельными молекулами.
Межмолекулярные взаимодействия делятся на:
– ориентационные – возникают между полярными молекулами.
При сближении полярных молекул они ориентируются таким образом, чтобы положительная сторона одного диполя была ориентирована к отрицательной стороне другого диполя (рисунок 15.1).
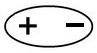
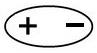
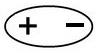
Рисунок 15.1– Ориентационное взаимодействие | ||||
– индукционные – возникают между полярными и неполярными молекулами. Полярные молекулы индуцируют на неполярных диполи, которые затем электростатически взаимодействуют друг с другом (рисунок 15.2).
Рисунок 15.2 – Индукционное взаимодействие
– дисперсионные – возникают между неполярными молекулами. В любой неполярной молекуле или атоме благородного газа возникают флуктуации электрической плотности, в результате чего появляются мгновенные диполи, которые в свою очередь индуцируют диполи у соседних молекул. Образовавшиеся диполи взаимодействуют (рисунок 15.3).

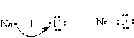





















| |||||||||||
Рисунок 15.3 – Дисперсионное взаимодействие | |||||||||||






Часть вторая. ХИМИЯ ЭЛЕМЕНТОВ
16 s-Элементы
s-Элементы – это элементы, у которых происходит заполнение s-подуровня. Данные элементы находятся в главных подгруппах первой и второй групп. S-элементы первой группы включают водород и щелочные металлы, а второй группы – бериллий, магний и щелочноземельные металлы. К s-элементам также относится инертный газ гелий.
Молекулярно-кинетическая теория — Википедия
Молекулярно-кинетическая теория (сокращённо МКТ) — теория, возникшая в XIX веке и рассматривающая строение вещества, в основном газов, с точки зрения трёх основных приближенно верных положений:
МКТ стала одной из самых успешных физических теорий и была подтверждена целым рядом опытных фактов. Основными доказательствами положений МКТ стали:
На основе МКТ развит целый ряд разделов современной физики, в частности, физическая кинетика и статистическая механика. В этих разделах физики изучаются не только молекулярные (атомные или ионные) системы, находящиеся не только в «тепловом» движении, и взаимодействующие не только через абсолютно упругие столкновения. Термин же молекулярно-кинетическая теория в современной теоретической физике уже практически не используется, хотя он встречается в учебниках по курсу общей физики.
Началом становления МКТ послужила теория М. В. Ломоносова[1][2]. Ломоносов опытным путём опроверг теории о теплороде и флогистоне, подготовив тем самым, молекулярно-кинетическую теорию XIX века Рудольфа Клаузиуса, Людвига Больцмана и Джеймса Максвелла.
p=13m0nv2{\displaystyle p={\frac {1}{3}}m_{0}nv^{2}}
Основное уравнение МКТ связывает макроскопические параметры (давление, объём, температура) газовой системы с микроскопическими (масса молекул, средняя скорость их движения).
Вывод основного уравнения МКТ[править | править код]
Пусть имеется кубический сосуд с ребром длиной l{\displaystyle l} и одна частица массой m{\displaystyle m} в нём.
Обозначим скорость движения vx{\displaystyle v_{x}}, тогда перед столкновением со стенкой сосуда импульс частицы равен mvx{\displaystyle mv_{x}}, а после — −mvx{\displaystyle -mv_{x}}, поэтому стенке передается импульс p=2mvx{\displaystyle p=2mv_{x}}. Время, через которое частица сталкивается с одной и той же стенкой, равно t=2lvx{\displaystyle t={\frac {2l}{v_{x}}}}.
Отсюда следует:
- Fx=pt=2mvx22l{\displaystyle F_{x}={\frac {p}{t}}={\frac {2mv_{x}^{2}}{2l}}}
Так как давление p=FS{\displaystyle p={\frac {F}{S}}}, следовательно сила F=p∗S{\displaystyle F=p*S}
Подставив, получим: pxS=mvx2l{\displaystyle p_{x}S={\frac {mv_{x}^{2}}{l}}}
Преобразовав: px=mvx2lS{\displaystyle p_{x}={\frac {mv_{x}^{2}}{lS}}}
Так как рассматривается кубический сосуд, то V=Sl{\displaystyle V=Sl}
Отсюда:
px=mvx2V{\displaystyle p_{x}={\frac {mv_{x}^{2}}{V}}}.
Соответственно, py=mvy2V{\displaystyle p_{y}={\frac {mv_{y}^{2}}{V}}} и pz=mvz2V{\displaystyle p_{z}={\frac {mv_{z}^{2}}{V}}}.
Таким образом, для большого числа частиц верно следующее: Px=Nmvx2¯V{\displaystyle P_{x}=N{\frac {m{\bar {v_{x}^{2}}}}{V}}}, аналогично для осей y и z.
Поскольку v2=vx2+vy2+vz2{\displaystyle v^{2}=v_{x}^{2}+v_{y}^{2}+v_{z}^{2}}, то vx2¯=vy2¯=vz2¯=13v2¯{\displaystyle {\bar {v_{x}^{2}}}={\bar {v_{y}^{2}}}={\bar {v_{z}^{2}}}={\frac {1}{3}}{\bar {v^{2}}}}. Это следует из того, что все направления движения молекул в хаотичной среде равновероятны.
Отсюда Px=Py=Pz=P=Nmv2¯3V{\displaystyle P_{x}=P_{y}=P_{z}=P={\frac {Nm{\bar {v^{2}}}}{3V}}}
или PV=N3mv2¯{\displaystyle PV={\frac {N}{3}}m{\bar {v^{2}}}}.
Пусть Ek{\displaystyle \,E_{k}} — среднее значение кинетической энергии одной молекулы, тогда:
PV=23NEk=νRT{\displaystyle PV={\frac {2}{3}}NE_{k}={\nu }RT}, откуда, используя то, что ν=NNA{\displaystyle {\nu }={\frac {N}{N_{A}}}}(количество вещества), а R=NAk{\displaystyle R=N_{A}k}, имеем Ek=32kT{\displaystyle {E_{k}}={\frac {3}{2}}kT}.
Уравнение среднеквадратичной скорости молекулы[править | править код]
Уравнение среднеквадратичной скорости молекулы легко выводится из основного уравнения МКТ для одного моля газа.
Ek=12mv2¯=32kT{\displaystyle E_{k}={\frac {1}{2}}m{\bar {v^{2}}}={\frac {3}{2}}kT},
Nam=Mr{\displaystyle N_{a}m=M_{r}}, где Mr{\displaystyle M_{r}} — молярная масса газа, m{\displaystyle m} — масса молекулы газа.
Отсюда окончательно
v¯=3kTNAMr=3kTm{\displaystyle {\bar {v}}={\sqrt {\frac {3kTN_{A}}{M_{r}}}}={\sqrt {\frac {3kT}{m}}}}[3]
- ↑ Фигуровский Н. А. Очерк общей истории химии. От древнейших времен до начала XIX в. — М.: Наука, 1969
- ↑ Михаил Васильевич Ломоносов. Избранные произведения в 2-х томах. М.: Наука. 1986
- ↑ Сивухин Д. В. Термодинамика и молекулярная физика // Общий курс физики. — М.: Наука, 1975. — Т. II. — С. 258. — 38 000 экз.
Биомолекулы — Википедия
Биомолекулы — это органические вещества, которые синтезируются живыми организмами. В состав биомолекул включают белки, полисахариды, нуклеиновые кислоты, а также более мелкие компоненты обмена веществ. Биомолекулы состоят из атомов углерода, водорода, азота, кислорода, а также фосфора и серы. Другие атомы входят в состав биологически значимых веществ значительно реже.
Среди биомолекул выделяют:
- Малые молекулы:
- Мономеры, олигомеры и полимеры
Нуклеозиды образуются при присоединении азотистого основания к сахару рибозе, примерами нуклеозидов являются цитидин, уридин, аденозин, гуанозин, тимидин и инозин.
Нуклеозиды в клетках могут быть фосфорилированы киназами, при этом образуются нуклеотиды. ДНК и РНК являются линейными полимерами, состоящими из относительно низкомолекулярных мономеров — нуклеотидов, соединенных между собой фосфодиэфирными связями.[1]
Нуклеотиды могут быть источниками энергии, запасенной в химических связях (АТР), принимать участие в передаче сигнала внутри клетки (cGMP, cAMP), являться компонентами кофакторов ферментов (кофермент А, FAD, NAD).[2]
Моносахариды — простейшие углеводы, обычно содержат альдегидную или кето-группу.[3] Наличие в структуре альдегидной группы обозначается приставкой «альдо-«, а кето-группы — «кето-«.[1] Примерами моносахаридов являются гексозы — глюкоза, фруктоза, галактоза и пентозы — рибоза и дезоксирибоза.[3]
Дисахариды образуются при соединении двух молекул простых сахаров, при этом отщепляется одна молекула воды. Дисахариды могут быть гидролизованы до соответствующих моносахаридов разбавленными растворами кислот или соответствующими ферментами.[1] Представителями дисахаридов являются сахароза, мальтоза и лактоза.
Полисахариды являются сложными сахарами, полимерами моносахаридов. Представителями полисахаридов является крахмал, целлюлоза и гликоген. Молекулы полисахаридов обычно имеют разветвленную структуру. Как правило, полисахариды нерастворимы или малорастворимы в воде, однако может происходить гидратация их гидроксильных групп, в таком случае при нагревании в водной среде полисахарид образует коллоид.[1] Более короткие полисахариды, состоящие из 2-10 мономеров, называют олигосахаридами.[4]
Лигнин — это нерегулярный биополимер, состоящий из ароматических колец, соединенных короткими (от одного до трех атомов углерода) углеродными цепями. Лигнин является вторым по значению биополимером после целлюлозы, и является одним из структурных компонентов растений.[5] Лигнин является рацематом, то есть не обладает оптической активностью, не поляризует свет. Эта особенность лигнина вызвана тем, что его полимеризация происходит по свободно-радикальному механизму.
Липиды в основном представлены сложными эфирами жирных кислот и являются важными компонентами клеточных мембран. Также липиды выполняют функцию запаса энергии, например, триглицериды. Большинство молекул липидов состоит из гидрофильной головки и от одного до трех гидрофобных хвостов жирных кислот, поэтому липиды являются амфифильными веществами.
В клеточных мембранах представлены следующие классы липидов:
Также к липидам относят простагландины и лейкотриены, 20-углеродные молекулы, синтезируемые из арахидоновой кислоты.
Аминокислоты содержат амино- и карбоксильную группу и являются цвиттер-ионами. Биологически значимые аминокислоты представлены только α-аминокислотами, в которых функциональные группы соединены с одним атомом углерода, а также пролином, который является иминокислотой.
Аминокислоты являются мономерами пептидов (2-10 остатков аминокислот), полипептидов и белков. Белки выполняют различные функции в клетке.
Биологически значимы только 20 аминокислот, они закодированы в генетическом коде, всего известно более пятисот природных аминокислот. Известны как минимум две аминокислоты, которые также встраиваются в полипептиды в ходе трансляции у некоторых организмов:
Другие биологически значимые аминокислоты представлены в том числе карнитином, орнитином, гамма-аминомасляной кислотой и таурином.
Витамины — вещества, которые организм не способен синтезировать самостоятельно, но необходимые для жизнедеятельности. Витаминами являются, например, многие коферменты. Витамины должны поступать в организм постоянно, обычно в очень малых количествах.
- ↑ 1 2 3 4 Slabaugh, Michael R., and Seager, Spencer L. Organic and Biochemistry for Today (неопр.). — 6th. — Pacific Grove: Brooks Cole (англ.)русск., 2007. — ISBN 0-495-11280-1.
- ↑ Alberts B., Johnson A., Lewis J., Raff M., Roberts K & Wlater P. Molecular biology of the cell (неопр.). — 4th. — New York: Garland Science (англ.)русск., 2002. — С. 120—121. — ISBN 0-8153-3218-1.
- ↑ 1 2 Peng, Bo, and Yu Qin. Fructose and Satiety (англ.) // Journal of Nutrition (англ.)русск. : journal. — 2009. — June. — P. 6137—6142.
- ↑ Pigman, W.; D. Horton. The Carbohydrates (неопр.). — San Diego: Academic Press, 1972. — Т. 1A. — С. 3. — ISBN 68-26647.
- ↑ K. Freudenberg & A.C. Nash (eds). Constitution and Biosynthesis of Lignin (неопр.). — Berlin: Springer-Verlag, 1968.
Межзвёздные молекулы — Википедия
Материал из Википедии — свободной энциклопедии
Межзвездные молекулы — молекулы, обнаруженные в межзвездной среде.
Двухатомные молекулы (СН, СН+, CN) в межзвездной среде были найдены оптическими методами в конце 30-х годов XX века по линиям поглощения в спектрах звёзд. Долгое время возможность существования в межзвездной среде молекул с числом атомов более двух считалась маловероятной. Первая многоатомная молекула, молекула аммиака (NH3), в межзвездной среде была открыта группой Ч. Таунса в 1968 году. Первая органическая молекула, формальдегид (Н2СО), была обнаружена в 1969 году. Подавляющее большинство молекул в межзвездной среде было открыто методами радиоастрономии (вращательная и вращательно-колебательная спектроскопия). В настоящее время надежно отождествлено около 100 видов межзвездных молекул с учётом молекул различного изотопного состава, в том числе, большое количество органических молекул, содержащих до 70 атомов. В настоящее время наиболее тяжелой многоатомной молекулой является обнаруженная в межзвездной среде в 2010 году молекула фуллерена, состоящая из 70 атомов.
Особенностью молекулярного состава наиболее плотных молекулярных облаков является преобладание в них органических соединений. Обнаружены представители нескольких классов органических соединений — альдегидов, спиртов, простых и сложных эфиров, карбоновых кислот, амидов кислот. Неожиданным было обнаружение в межзвездной среде относительно сложных многоатомных молекул. Многие из этих соединений (HCN, CH2NH, СН3NН2 и др.) известны как активный исходный материал для образования важнейших предбиологических молекул — аминокислот и азотистых оснований. Это является важным аргументом в пользу универсальности путей химической эволюции во Вселенной.
Межзвездные молекулы с 10 и более атомами[править | править код]
- Стрельницкий В. С. Межзвездные молекулы. М.: Знание, 1974.
- Woon, David E. Interstellar and Circumstellar Molecules (неопр.) (October 1, 2010). Дата обращения 4 октября 2010. Архивировано 2 февраля 2013 года.
- Molecules in Space (неопр.) (недоступная ссылка). Universität zu Köln (August 2010). Дата обращения 4 октября 2010. Архивировано 2 февраля 2013 года.
- Dworkin, Jason P. Interstellar Molecules (неопр.). NASA’s Cosmic Ice Lab (February 1, 2007). Дата обращения 23 декабря 2010. Архивировано 2 февраля 2013 года.
- Wootten, Al The 129 reported interstellar and circumstellar molecules (неопр.). National Radio Astronomy Observatory (November, 2005). Дата обращения 13 февраля 2007. Архивировано 2 февраля 2013 года.
- Lovas, F. J.; Dragoset, R. A. NIST Recommended Rest Frequencies for Observed Interstellar Molecular Microwave Transitions, 2002 Revision (неопр.) (недоступная ссылка). National Institute of Standards and Technology (February, 2004). Дата обращения 13 февраля 2007. Архивировано 2 февраля 2013 года.